
Abstract
Very-low-density lipoproteins (VLDL) is a hallmark of metabolic syndrome (MetS) and each manifestation of MetS is related to atrial fibrillation (AF) risks. Slowed atrial conduction is a mechanism of AF in MetS. We hypothesized that VLDL can modulate and reduce atrial gap junctions. VLDLs were separated from normal (Normal-VLDL) and MetS (MetS-VLDL) individuals. VLDLs (15 µg/g) and equivalent volumes of saline (CTL) were injected respectively to C57BL/6 mice for 6 weeks. Electrocardiograms demonstrated that MetS-VLDL induced prolongation of P wave (P = 0.041), PR intervals (P = 0.014), QRS duration and QTc interval (both P = 0.003), but Normal-VLDL did not. Optical mapping of perfused hearts confirmed slowed conduction on atria and ventricles of MetS-VLDL mice. Slowed cardiac conduction was associated with significant atrial and ventricular remodeling, along with systolic dysfunction and comparable intra-cardiac fibrosis. MetS-VLDL induced downregulation of Cx40 and Cx43 at transcriptional, translational and tissue levels, and it also enhanced O-GlcNAcylation of Cx40 and Cx43. Protein structure analyses predicted O-GlcNAcylation at serine 18 of Cx40 and Cx43 which may impair stability of gap junctions. In conclusion, MetS-VLDL modulates gap junctions and delays both atrial and ventricular conduction. VLDL may contribute to the pathophysiology of atrial fibrillation and ventricular arrhythmias in MetS.
Similar content being viewed by others

Introduction
Metabolic syndrome (MetS) is not only an important risk factor of atrial fibrillation (AF) but also increases the morbidity and mortality for patients with AF. For aging population, the incidence of AF is increasing, along with its complications such as thromboembolism, stroke, and heart failure causing disabilities, morbidity and mortality1. Despite decades of extensive researches, prevention of AF remains a challenge.
One of the key components of MetS is dyslipidemia which manifests with reduced high-density lipoprotein and elevated triglycerides, which is mostly carried by very-low density lipoprotein (VLDL) in blood circulation. In our previous study, VLDL of individuals with MetS (MetS-VLDL) was found different from VLDL of healthy individuals (Normal-VLDL), in terms of abundance of electronegativity and apolipoprotein C-rich fraction. MetS-VLDL exerts substantially larger internalization and cytotoxicity to endothelial cells and atrial myocytes2,3. In vivo, MetS-VLDL induces atrial remodeling and vulnerability of AF in our VLDL-injection mouse model3. The mechanisms warrant further studies.
Electrical conduction occurring along atria from the sinus node to the ventricles can be reflected by the PR interval in electrocardiography. The Framingham heart study has defined prolonged PR interval as a risk factor for AF4. PR prolongation is also associated with adverse outcomes such as incidence of pacemaker implantation, and all-cause mortality5,6. Clinical studies suggest that PR interval prolongation is related to obesity, increased waist circumference, and components of MetS7. Electrophysiological causes of PR interval prolongation include intra-atrial conduction defects, conduction disease in the AV node, conduction disease in the His Bundle, increased vagal tone and the presence of medicine leading to increased AV nodal conduction delay8. In addition to PR interval, prolongation of P wave duration was also found robustly associated with an increased risk of AF in a large primary care population9. The mechanisms underlying atrial conduction defect in MetS remain unclear.
Accumulating evidence in animal studies suggests that there is a link between gap junction changes and AF10,11,12,13,14. The gap junction proteins Cx40 and Cx43 that are encoded by gene GJA5 and GJA1 respectively, these together form the low resistance pathways for electrical activity between atrial cardiomyocytes and are responsible for coordinated electrical activity in the atria. Changes in Cx40 expression and distribution in the atrial myocardium have been shown in parallel with reduced atrial effective refractory period, enhanced spatial dispersion of atrial refractoriness, or abnormal atrial impulse conduction15. In addition, GJA5 mutations or polymorphisms bring an inherited predisposition to AF16.
Post-translational modifications are also important for connexins regulation17. The O-GlcNAcylation of connexins is a posttranslational, and reversible modification that involves the addition of N-acetylglucosamine (GlcNAc) on serine or threonine residues. O-GlcNAcylation can also regulate the phosphorylation of Cx40 and Cx43. Evidence has shown a contribution of O-GlcNAcylation to endothelial dysfunction in diabetes mellitus and in hyperglycemia-associated cardiovascular defects18,19. The O-GlcNAcylation of Cx40 or Cx43 in atrial myocytes has not been explored.
The modulation of gap junctions on the occurrence of AF has not been examined in the MetS model. In our VLDL-injection mouse model, the atrial remodeling and AF susceptibility to sympathetic stimulation are observed with injection of MetS-VLDL, but not with injection of Normal-VLDL3. Using this model, the study was aimed to examine the hypothesis that MetS-VLDL can modulate atrial connexins expression and slow atrial conduction. Whether MetS-VLDL induces O-GlcNAcylation of Cx40 and/or Cx43 in atrial myocytes and whether O-GlcNAcylation would change the gap junction stability were also assessed. The results are of general importance with respect to pathogenesis of AF in MetS.
Results
In vivo MetS-VLDL resulted in prolonged P waves, PR intervals, QRS width, and QT intervals
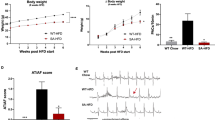
To determine the in vivo effect of VLDL on intra-cardiac electrical conduction, we used surface ECG to determine parameters of P wave width, PR intervals, QRS width, and QT intervals of three groups mice with either saline (control), Normal-VLDL (nVLDL), and MetS-VLDL injection (msVLDL) (Fig. 1a and b, Table 1). Similar to human ECG, the murine ECG shows P waves, reflecting spread of atrial depolarization from impulse of sinoatrial node. Subsequently, the PR interval reflects impulse conduction from the atria through the AV node and the His-bundle to the ventricles. Then the QRS complex reflects ventricular depolarization. The QT interval was defined as the period between the initiation of the Q wave to the point of T waves returning to the isoelectrical baseline. The murine QTc interval is corrected with heart rate using Bazett’s formula, which is commonly used in human ECG analysis20. In msVLDL mice (n = 10), P wave was significantly wider than nVLDL (n = 10) and control mice (n = 10) (Control 19.4 ± 6.4 vs nVLDL 19.3 ± 5.6 vs msVLDL 27.8 ± 12.3 msec, ANOVA P = 0.041; $p = 0.01, #p = 0.03; Fig. 1b). The PR interval was significantly prolonged in msVLDL mice (Control 47.4 ± 12.2 vs nVLDL 54.3 ± 10.6 vs msVLDL 71.4 ± 16.8 msec, ANOVA P = 0.015; $P < 0.01, #P = 0.03; Fig. 1b). The msVLDL also had wider QRS ($P < 0.05, #P < 0.01; Fig. 1b) and QT prolongation ($P < 0.01, #P < 0.01; Fig. 1b). In summary, these findings indicate that MetS-VLDL delayed intra-atrial, atria-ventricular, and ventricular conduction and ventricular repolarization. Moreover, unprovoked AF was only observed in msVLDL mice after 6 week’s MetS-VLDL injection and before sacrifice (Fig. 1c).

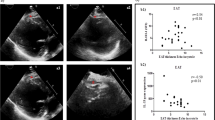
Prolongation of P waves, PR intervals, QRS, and QT intervals along with Left atrial and left ventricular dilatation and comparable intra-cardiac fibrosis. (a) The dash lines indicate measurements of P wave width (P), PR interval (PR), QRS width, and QT interval. (b) P, PR, QRS, and QTc in MetS-VLDL injected mice (msVLDL), Normal-VLDL injected mice (nVLDL) and control mice (n = 10 for each) $P < 0.05; $P < 0.05. (c) Representative electrocardiographic tracings of unprovoked atrial fibrillation from two msVLDL mice. (d) B-mode and M-mode images for measurements of dimensions of aortic root (AO), left atria (LA), and left ventricles (LV). (e) LA size and LV end-diastolic dimension (msVLDL vs control, $P < 0.01; msVLDL vs nVLDL, #P < 0.01). (f) Gross morphology of hearts. The msVLDL atrium was larger than the control and nVLDL ones. (g) Histological sections of control, nVLDL, and msVLDL stained with Masson-Trichrome; black bars indicate 50 μm.
MetS-VLDL induced left atrial and ventricular dilation and impaired left ventricular systolic function without enhancing cardiac fibrosis
The cardiac structure and left ventricular function were examined by echocardiography after 6 week’s VLDL injection (Fig. 1d and e, and Table 2). Although both VLDLs seemed to increase LV mass (ANOVA P = 0.4372), only MetS-VLDL induced significant dilatation of left atria (P < 0.0001 vs control and P < 0.0001 vs nVLDL) and left ventricles (P = 0.0189 vs control). The dilated left ventricular diameter and volume in msVLDL mice hearts were associated with reduced left ventricular ejection fraction (EF, msVLDL 60.3 ± 7.3% vs control 67.6 ± 7.9%; P = 0.0086) and fraction shortening (FS, msVLDL 32.0 ± 4.9% vs 37.4 ± 6.2%; P = 0.0096). These results indicate that MetS-VLDL induced significant atrial and ventricular remodeling along with impaired left ventricular systolic functions.
After hearts were dissected and washed, the atria and ventricles were grossly larger for the msVLDL groups (Fig. 1f). Increased fibrosis in myocardium can slow myocardial conduction. Therefore, Mason-Trichrome staining of the atrial and ventricular tissues was performed and the fibrosis region was quantified by ImageJ software. Degree of fibrosis was comparable among groups (Fig. 1g). Therefore, the delayed atrial and ventricular conduction in msVLDL mice is not related to intra-cardiac fibrosis.
In vivo downregulation of Cx40 and Cx43 mRNA and protein induced by MetS-VLDL
Decreased gap junction function can potentially slow myocardial conduction. Therefore, we examine if expression of cardiac connexins can be reduced by MetS-VLDL. Atrial and ventricular expression of GJA5, and GJA1 mRNA were determined by real-time PCR (Fig. 2a). Protein levels of Cx40 and Cx43 were determined by Western blotting (Fig. 2b). For atria, GJA5 and GJA1 mRNA was decreased in msVLDL (0.083 ± 0.062 and 0.127 ± 0.096 folds to control, n = 3), nonsignificant when compared to nVLDL (0.530 ± 0.421 and 0.561 ± 0.436 folds to control, n = 3; P = 0.143 and P = 0.168) and significantly compared to control mice (msVLDL vs conrtol, P = 0.0028 for GJA5, P = 0.014 for GJA1). For ventricles, GJA5 and GJA1 mRNA was significantly decreased in msVLDL (0.352 ± 0.155 and 0.207 ± 0.044 folds to control, n = 5), compared to nVLDL (1.043 ± 0.225 and 1.069 ± 0.351 folds to control, n = 5; P = 0.0005 and P = 0.0006), and control (P = 0.0003 and P < 0.0001). The protein levels of Cx40 and Cx43 were significantly decreased in msVLDL atria (0.60 ± 0.17 and 0.40 ± 0.08 folds to control, n = 4), compared to nVLDL group (0.90 ± 0.11 and 0.72 ± 0.04 folds to control, n = 4; P = 0.023 and P = 0.0004), and the control (P = 0.004 and P < 0.0001). In ventricles, the protein levels of Cx40 and Cx43 were also significantly decreased in msVLDL mice (0.60 ± 0.11 and 0.82 ± 0.04 folds to control, n = 4), compared to nVLDL group (0.99 ± 0.09 and 1.05 ± 0.11 folds to control, n = 4; P = 0.0015 and P = 0.01), and the control (P = 0.0067 and P = 0.0045). In vivo effect of VLDLs on atrial expression of Cx40 and Cx43 was further assessed by the immunohistochemistry study (Fig. 2c and d). In msVLDL atria, Cx40 and Cx43 were significantly down-regulated (0.376 ± 0.164 and 0.403 ± 0.285 folds to control, P = 0.043 and P = 0.044 vs control); whereas Cx40 and Cx43 changes in nVLDL were nonsignificant (0.486 ± 0.227 and 0.740 ± 0.104 folds to control, P = 0.165 and P = 0.164 vs control). These findings indicate that MetS-VLDL substantially reduced atrial Cx40 and Cx43 expression at transcriptional, translational, and tissue levels, suggesting the pathogenic role of Cx40 and Cx43 reduction on delayed atrial conduction and development of AF in msVLDL mice.

The transcriptional, translational and tissue expression of Cx40 and Cx43. (a) Results of real-time PCR of GJA5 and GJA1 in atrial and ventricular tissues. For atria (n = 3 for each), GJA5 $P = 0.0028 and GJA1 $P = 0.014 for GJA1; msVLDL vs control. For ventricles (n = 5 for each), GJA5 #P = 0.0005, msVLDL vs nVLDL; $P = 0.0003, msVLDL vs control; GJA1 #P = 0.0006 and $P < 0.0001). (b) Western blotting for atrial and ventricular expression of Cx40 and Cx43. For atria Cx40 (n = 4 for each), msVLDL vs control, $P = 0.004; msVLDL vs nVLDL, #P = 0.023. For atria Cx43 (n = 4 for each), msVLDL vs control, $P < 0.0001; msVLDL vs nVLDL, #P = 0.0004; nVLDL vs control, *P = 0.0003. In msVLDL ventricles, Cx40 and Cx43 protein reduction is significant (#P = 0.0015 and #P = 0.01 vs nVLDL; $P = 0.0067 and $P = 0.0045 vs control). (c) Immunohistochemistry of atrial tissues (n = 7 for each). Nuclei with DAPI staining appear blue. The plasma membrane with WGA staining appear red. Cx40 (upper panel) and Cx43 (lower panel) appear green. The scale bars indicate 20 µm. (d) The quantification of Cx40 and Cx43 signaling integrated from a series of confocal image sections (Cx40 and Cx43 integration). For msVLDL, Cx40 and Cx43 (0.376 ± 0.164 and 0.403 ± 0.285 folds to control) are significantly reduced ($P = 0.044 and $P = 0.043 vs control).
MetS-VLDL induced delayed atrial and ventricular conduction
To evaluate if reduced Cx40 and Cx43 co-exist with slower intra-cardiac conduction velocity, the optical mapping technique was used to image the membrane potential of intact atria and ventricles on ex-vivo perfused hearts21. The membrane potential propagations and the derived conduction velocity of CTL, nVLDL, and msVLDL ex-vivo hearts during rapid atrial pacing (pacing cycle length = 200 ms) were demonstrated in the Fig. 3. The conduction velocity in atria and ventricles were all slower in msVLDL compared with CTL and nVLDL mice (right atria P = 0.0123; left atria, P = 0.0222; right ventricles P = 0.0091; left ventricle, P = 0.0004 by ANOVA analysis).

Atrial and ventricular conduction evaluated by optical mapping in CTL, nVLDL, and msVLDL mice. (a) The right atrial propagations in CTL, nVLDL, and msVLDL during rapid atrial pacing (pacing cycle length = 200 ms) were demonstrated in the isochronal map. The white asterisk indicates the pacing site. (b) The conduction velocity was compared among CTL, nVLDL, and msVLDL groups. The conduction velocity in RA, RV and LV were all slower in msVLDL than in CTL and nVLDL mice. Although the conduction velocity in LA is slower in msVLDL than in nVLDL mice, there is no difference in conduction velocity comparing LA in msVLDL than CLT group. $P < 0.05 compared with CTL. #P < 0.05 compared with nVLDL.
O-GlcNAcylation of Cx40 and Cx43 in atrial myocytes was induced by MetS-VLDL but not by Normal-VLDL
It is known from Cx40+/− mice that the PR interval and atrial conduction velocity is not delayed with a 50% reduction of Cx40 protein expression22,23. Except for the expression level, posttranslational modification plays a critical role in determining gap junction function. We hypothesized that MetS-VLDL can induce O-GlcNAcylation of Cx40 or Cx43 in cardiomyocytes. Atrial myocytes HL-1 were cultured and treated with either MetS-VLDL or Normal-VLDL of 25 mg/L for 16 hours. The immunoprecipitation revealed that Cx40 and Cx43 were markedly O-GlcNAcylated in the MetS-VLDL group (Cx40, P = 0.0313 vs control; Cx43, P = 0.017 vs control) (Fig. 4); whereas Normal-VLDL did not increase O-GlcNAcylation of Cx40 and Cx43 at the same concentration with the same duration of treatment.

The MetS-VLDL induced O-GlcNAcylation of Cx40 and Cx43. (a) The immunoblot shows increase in total O-GlcNAcylated Cx40 and Cx40 proteins in MetS-VLDL group. (b) Densitometry demonstrates that in the MetS-VLDL group, a significant increase in O-GlcNAcylation levels for both Cx40 and Cx43 (n = 4 for each group; 1.37 ± 0.26 folds to control, $P = 0.03 for Cx40 and 1.36 ± 0.15 folds to control, $P = 0.02 for Cx43).
Molecular modeling and structural analysis of human Cx40 and Cx43
Next, the computational modeling was used to test the hypothesis that O-GlcNAcylation modification of Cx40 and Cx43 can affect the gap junction stability. Firstly, human Cx40 and Cx43 oligomerization models were constructed using the published crystal structure Cx26 gap junction (PDB code 2ZW3) as the template (Fig. 5). As the result, connexins oligomerize into hexamers and the connexin was predicted to have four trans-membrane helices and two extracellular loops, which are thought to contain a β-strand structure and are an essential structural basis for the docking of two connexons to form the complete gap junction channel24. Then the O-GlcNAcylation site prediction was performed. Both sequence-based (OGTSite) and structure-based (Glycoprotein Builder) analyses predicted the most possible O-GlcNAcylation sites of Cx40 and Cx43 at the residue Serine 18, which is also located at the interface between the connexins. This finding suggested that the O-GlcNAcylation of Serine 18 may disturb stability between connexins interface and thus hamper the oligomerization of connexins into connexons.

Structure of the O-GlcNAcylated Cx40 and Cx43 proteins and their electrostatic potential surface. (a) The side view of Cx40 structure with O-GlcNAcylation at the residue Serine 18. (b) The top view of Cx40 with O-GlcNAcylation. (c,d) The electrostatic potential surface of Cx40 and Cx43 with O-GlcNAcylation. The O-GlcNAc on the surface is highlighted with dotted circles. Electrostatic potential surfaces are blue for positive, white for hydrophobic, and red for negative. All images were generated by the PyMOL (Available: http://www.pymol.org).
Discussion
The key findings of this study are as follows: (1) MetS-VLDL can significantly reduce Cx40 and Cx43 mRNA and protein expression in atrial and ventricular tissues. (2) MetS-VLDL can induce modification of O-GlcNAcylation on Cx40 and Cx43 connexins. (3) MetS-VLDL slowed atrial and ventricular conduction, and delay ventricular repolarization; whereas Normal-VLDL did not exert the aforementioned findings. (4) The O-GlcNAcylation of Cx40 was predicted at the residue Serine 18, and as a result the stability between adjacent connexins binding may be disturbed. Noteworthy, the delayed cardiac conduction in msVLDL mice is concomitant with significant atrial and left ventricular remodeling and systolic dysfunction, whereas are not related to intra-cardiac fibrosis. This study is extended from our previous works that identified remarkable differences between MetS-VLDL and Normal-VLDL on inducing AF vulnerability2,3, and the key findings may further explain early pathogenesis of AF in MetS.
Cardiac electrophysiology in mouse models has been well validated. The normal range for mice PR interval is 30–56 msec and for QRS duration is 9–30 msec20. In this study, values for the control were consistent and within the normal ranges. Mice with MetS-VLDL injection had values longer than the normal limits, but those with Normal-VLDL did not. Similarly, QT and QTc intervals were prolonged in msVLDL mice and were normal in control and nVLDL mice which were consistent to previous studies20,25,26. Also, consistent with a recent delicate work using high-fat-diet mice by Takahashi et al., prolonged P wave duration was also observed and it was shown associated with downregulation of Cx40 and atrial tachycardia inducibility27. However, PR interval and QRS duration in their high-fat-diet mouse model were not prolonged.
Cx43 is expressed in cardiomyocytes throughout the heart, whereas Cx40 expression is exclusively to the atria and the His bundle and Purkinje fibers28. Deficiencies in either Cx43 or Cx40 result in cardiac conduction defects28. For atrial tachyarrhythmia, Cx40 downregulation was suggested more important than Cx4311,29. Studies have shown that P wave duration is not affected with reduced Cx43 levels in Cx43+/− mice30. Re-distribution of Cx43, in which Cx43 is moved from the primary location of intercalated disc between adjacent cardiomyocytes to disperse throughout the sarcolemma, has been suggested more closely related to cardiac conduction delay11,31. For Cx40, conflicting results have been reported with respect to the effect of Cx40 deficiency on atrial conduction29. With a 50% reduction of Cx40 protein in Cx40+/− mice, PR interval and atrial conduction velocity was not significantly changed22,23. The aforementioned works help to explain why nVLDL mice, in which with an approximate 30% reduction of atrial Cx40 expression, had unaffected durations of P wave and PR intervals.
Oligomerization of connexins into hemichannel (the hexamer, connexon) takes place after the protein exits the endoplasmic reticulum. Once delivered to the cell surface, a pair of connexons from neighboring cells form gap junction plaques. The half-life of gap junctions is short (<5 h) and both proteasomal and lysosomal degradation of gap junctions are thought involved in connexin degradation32. O-GlcNAcylation of connexins may affect the assembly of connexons and resulting change of gap junction turnover. In endothelial cells under high-glucose condition, and O-GlcNAcylated Cx40 was shown to cause gap junction dysfunction and resulted in endothelial dysfunction19. It is possible that O-GlcNAcylation directly affects gating of gap junction channels or indirectly through competing for a phosphorylation of adjacent residues, and/or impairs stability of gap junction structure. To our knowledge, this study for the first time suggested an important role of O-GlcNAcylation for cardiac gap junctional function. We predicted the O-GlcNAcylated residues for Cx40 and Cx43. This prediction is crucial to determine further detail mechanism such as the biophysical impact of O-GlcNAcylated residue to the gap channel gating. Certainly, additional work is needed to fully establish the connection between Cx40 and Cx43 O-GlcNAcylation and MetS-VLDL-induced delayed atrial and ventricular conduction in this mouse model. The application of either the gap junctional coupling estimation with a dye transfer or the trans-junctional voltage-gating property of cells pairs measured on the dual whole-cell voltage-clamp technique, could shed some light on the impact of O-GlcNAcylation on Cx40 and Cx43 gap junctional function.
Although the regulation of circulatory VLDL concentration is complex, elevated VLDL is a hallmark of the MetS. A large-scale clinical study reported that circulatory lipoprotein subclasses including VLDL particles are modified in individuals with abnormal glucose tolerance and newly diagnosed diabetes mellitus33. On the other hand, another clinical study with mean follow-up of 9.6 years identified high-density lipoprotein cholesterol and triglycerides as independent risk factors for AF incidence34. Several clinical studies and meta-analysis consistently indicated a reduced risk of AF in patients treated with statins and the mechanisms of the beneficial effects remain uncertain35. These clinical evidences all indicate that circulatory VLDL may be crucial in AF pathogenesis. However, we could not find any report evaluating the correlation electrocardiography P wave or LA remodeling with circulatory VLDL in human beings. The unnoticed role of VLDL in MetS-related cardiovascular disease may be due to an overlook of differences between MetS-VLDL and Normal-VLDL. Further clinical and translational studies are required to elucidate the putative mechanism in which VLDL induces atrial and ventricular remodeling in MetS.
Upon our main research hypothesis for this study, the delayed ventricular conduction and repolarization in msVLDL mice were additional findings. Consistent with our previous study, significant cardiac remodeling became phenomenal after 6 week’s MetS-VLDL injection. Compared with our previous published data done with young mice (14-week-old), the aged mice (11-month-old) had larger atrial and ventricular sizes and larger ventricular mass3. In aged mice, MetS-VLDL significantly induced not only left atrial dilation, but also left ventricular dilation associated with reduced left ventricular ejection fraction. These observations suggest that MetS-VLDL exerts more cardiac lipotoxicity to aged mice than to young mice. From clinical observational studies, the association of QRS and QT prolongation with MetS has been noticed36,37. It is well known that prolonged QRS duration and QT prolongation are risk factors for sudden cardiac death. The adverse left ventricular remodeling, systolic dysfunction, and coexisting delayed ventricular conduction and repolarization in the msVLDL mice suggest an important role of VLDL in pathophysiology of ventricular arrhythmias in individuals with MetS. Whether our msVLDL mice are vulnerable to ventricular arrhythmias remains undetermined.
This study used human VLDL isolated from MetS patients and normal individuals to inject into immune-competent mice for 6 weeks. Although the VLDL and the VLDL receptor are similar and quite conserved in the human beings and mice, it is certainly that immune response was developed in these mice against foreign (human) along with the injection. The possibility that mice develop different immune responses on MetS-VLDL and Normal-VLDL cannot be excluded. In addition, some parameters of putative interest were unavailable, such as determination of VLDL particle size, VLDL levels after injection, and lipidomics of VLDL samples that were used in this study.
In conclusion, this study highlights evidence that MetS-VLDL in vivo and in vitro modulates cardiac gap junctions Cx40 and Cx43. Delayed atrial and ventricular conduction is induced by MetS-VLDL, at least in part, via its distinct effect on substantial decrease in Cx40 and Cx43 expression, and on enhancing the O-GlcNAcylation modification. We suggest that the MetS-VLDL plays a pivotal role in cardiac remodeling and pathogenesis of AF and ventricular arrhythmia. VLDL may be a novel target for prevention or control of AF, particularly as concerning individuals with MetS.
Methods
Human VLDL isolation
We followed the Helsinki Declaration principles. The research was approved by the Kaohsiung Medical University Hospital Ethics Review Board. Individuals who either met the criteria of the MetS or otherwise were healthy gave informed consent for blood donations. Then Normal-VLDL and MetS-VLDL (d = 0.930–1.006 g/mL) were isolated by sequential ultracentrifugation as previously described2,3,38.
Mice and diet
To test in vivo effects of VLDLs, we injected MetS-VLDL or Normal-VLDL intravenously to mice tail veins at a dose of 15 μg/g, three times a week for 6 consecutive weeks. The 15 μg/g dose was chosen to match the human normal VLDL concentration38, which is between 2 and 30 mg/dL. Nine-month-old C57BL/6 male mice from the National Laboratory Animal Center (Taipei, Taiwan) were maintained in a temperature-controlled facility (21 to 22 °C) with a 12-h light/dark cycle. The mice had free access to water and a standard chow diet. After a two-week acclimatization period, control and VLDL-injection groups were created by random separation of mice from the same lot. The control mice were injected with an equivalent 50 μL volume of phosphate buffered saline. Body weights were recorded weekly. Total number of mice used was 63 and all of them received saline or VLDLs injection, in which 18 were for the ex vivo optical mapping. All applicable institutional and governmental regulations concerning ethical use of animals were performed confirming to the NIH guidelines and all animal procedures were approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University.
Surface electrocardiography and echocardiography
Mice were anesthetized with 1.5–2% isofluorane. Platinum electrodes were inserted subcutaneously in the limbs and connected to a custom-built electrocardiogram (ECG) amplifier and recorded for about 5 minutes at 2 MHz. Raw tracings without clear P waves or T waves were discarded for intervals analysis. The analysis for intervals was done with LabChart 5 software (AD instruments), and QTc intervals were calculated with formula for mice: QTc = QT/(RR/100)1/2 25. In vivo heart chamber dimensions were measured for each mouse (control, n = 10; nVLDL, n = 10; msVLDL, n = 10) under anesthesiology with 1.5–2% isofluorane by using a Vevo2100 (VisualSonics, Inc., Toronto, Ontario, Canada) small animal instrument as previously described3.
Histology of mice atrial tissue
Mouse hearts were perfused with 4% paraformaldehyde at physiological pressures, blotted dry on tissue paper and weighed. Next, they were embedded in paraffin and sections were stained with Masson’s trichrome according to the manufacturer’s protocol as previously described. Measurements were performed using ImageJ software.
Confocal analysis of atrial tissue
Indirect immunofluorescent detection of Cx40, Cx43 was performed on frozen cross-sections of the atriums of mice of all experimental groups. Series of sections were fix in −20 °C cold methanol for 5 mins, blocked by 0.5% BSA in PBS and incubated with primary monoclonal antibody goat anti-40 (1:1000, Santa Cruz Biotechnology Inc. USA), rabbit anti-43 (1:1000, cell signaling technology, MA) and Wheat Germ Agglutinin, Texas Red®-X Conjugate (WGA, 1:500, Thermo Fisher Scientific Inc. USA) for 2 hours at 37 °C. The sections were subsequently washed with phosphate-buffered saline (PBS) and then followed the application of secondary anti-rabbit and anti-goat conjugated with FITC-fluorescein CF488A (1:500, Sigma-Aldrich Inc. USA) for 50 mins at room temperature. After washing, the sections were mounted into UltraCruz® Mounting Medium (with DAPI) and view by confocal microscope Olympus FV1000 (Olympus, Japan). Morphometric analyses were conducted on coronal sections using individual cell areas from atria that were traced and integrated with Olympus FV1000 software. In short, images were collected using the ×40 objective lens and zoom 1.0 computer setting so that each pixel represented 0.23 μm. Each image was consisted of 512 × 512 pixels. The projection view of 15 consecutive optical sections taken at interval of 0.53 μm was recorded for quantification analysis.
Quantitative real-time reverse transcriptase PCR and Western blot
Total RNA was prepared using TRI Reagent (Sigma-Adrich, St Louis, MO), then reverse transcribed (Invitrogen, Carlsbad, CA). Quantitative real-time RT-PCR was performed using an ABI 7500 real-time system (Applied Biosystems, Foster city, CA) and TaqMan Universal Master Mix II (Applied Biosystems, CA) with TaqMan probes GJA1 (Mm00439105_m1, Thermo Fisher Scientific Inc. USA) and GJA5 (Mm00433619_s1, Thermo Fisher Scientific Inc. USA). Protein samples were separated using a SDS-polyacrylamide gel and transferred to a nitrocellulose membrane.
Blots were incubated with anti-Cx40 (1:500 dilution; Santa Cruz), anti-Cx43 (1:1000 dilution; Cell Signal), and anti-phospho-Cx43 (1:1000 dilution; Cell Signal), then with horseradish peroxidase-linked secondary antibody. The immunoblots were identified with SuperSignal West Picochemiluminescent substrate (Thermo Fisher Scientific). Band intensity was calculated using ImageJ (National Institutes of Health). Intensity data from cytoplasmic and membraneous proteins in interest were normalized to α-tubulin and pan-cadherin, respectively.
Langendorff heart preparation
The mice were intraperitoneally injected with 200 units of heparin and anesthetized with sodium pentobarbital (35 mg/kg). After a median sternotomy, the hearts were harvested through a thoracotomy in ice-cold Tyrode’s solution (in mmol/L: NaCl 125, KCl 4.5, NaHCO3 24, NaH2PO4 1.8, CaCl2 1.3, MgCl2 0.5, and glucose 5.5) and cannulated via the aorta. The heart was then connected to a Langendorff apparatus and perfused by Tyrode’s solution with 95% O2 and 5% CO2 to maintain a pH of 7.35~7.45. A bipolar left atrial (LA) electrogram (LAE) and a pseudo-electrogram (p-ECG) were monitored. The p-ECG was obtained using widely spaced bipolar electrodes located in the bath surrounding the heart. The electrical signals were filtered from 0.05 to 100 Hz and digitized at 1 kHz using AxoScope (Molecular Devices, Downingtown, PA).
Optical mapping
Optical mapping techniques21 were used to study the Vm of intact heart. The perfused Tyrode’s solution was maintained at 37.5 °C with a constant flow rate ~2 ml/min. After 20 minutes of stabilization, the hearts were stained with 20 μL of the voltage-sensitive dye RH237 (2.5 mmol/L) for Vm mapping. The heart was then washed for 10 min followed by the addition of blebbistatin (15 mmol/L) (Tocris Bioscience, Minneapolis, MN). The stained hearts were illuminated with a LED at a 520-nm wavelength and the fluorescence was collected by a MiCAM05-Ultima CMOS camera (BrainVision, Tokyo, Japan) through a 715-nm long-pass filter. The fluorescence signal was recorded at 1 ms/frame in a 100 × 100 pixel grid with a spatial resolution of 0.06 × 0.06 mm2 per pixel. Optical signals were processed with both spatial (3 × 3 pixels Gaussian filter) and temporal (3 frames moving average) filtering. The fluorescence obtained through a common lens was separated with a dichroic mirror (650 nm cutoff wavelength), and directed to the respective camera with additional filtering (715 nm long pass for Vm).
HL-1 atrial myocyte culture
A murine HL-1 atrial myocyte cell line was maintained with fresh Claycomb medium in pre-coating culture flasks at 37 °C in a humidified atmosphere containing 5% CO2. When the cells reached confluence, splitting was performed by recommended passaging procedures. Culture medium was supplemented with 87% Claycomb medium, 2 mM/L L-glutamine, 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 0.1 mM/L norepinephrine. To measure effects of VLDLs on expression and O-GlcNAcylation of Cx40 and Cx43, HL-1 cells were seeded overnight and then treated with Normal-VLDL or MetS-VLDL at a concentration of 25 mg/L for 16 h (n = 4 for each group).
Detection of O-GlcNAcylated Cx40 and Cx43 proteins with immunoprecipitation
Protein extract was isolated from HL-1 cells using lysis buffer. Protein of 1 mg was mixed with anti-Cx40 (1:100 dilution, sc-20466, Santa Cruz) or anti-Cx43 (1:100 dilution, 15386-1-AP, Proteintech) at 4 °C for 1 h and then was incubated with Pierce™ Protein A/G Magnetic Beads (88802, Thermo Fisher Scientific) overnight at 4 °C. The IP matrix was washed twice by PBS containing 1% Nonidet P-40. The matrix-bound protein was eluted in sample buffer and then separation by 10% SDS-PAGE electrophoresis. The immunoblot was used the anti-OlcNAc (1:1000 dilution, MA1-076, Thermo Fisher Scientific), anti-Cx40 (1:500) or anti-Cx43 (1:1000) antibody overnight at 4 °C and signals was detected with secondary antibodies and chemiluminescence visualization.
Structure prediction of the O-GlcNAcylated Cx40 and Cx43
The three-dimensional structures of human Cx40 and Cx43 were predicted by using the MODELLER 9v839. For this study, the X-ray crystal structure of the Connexin-26 (Cx26) gap junction protein (PDB code 2ZW3) was selected as the template structure40. The models of Cx40 and Cx43 were built using this template. The O-GlcNAcylation sites of Cx40 and Cx43 were predicted by OGTSite41, which is a web server for identifying O-GlcNAcylation sites with their corresponding OGT substrate motifs. The structures of the O-GlcNAcylated Cx40 and Cx43 variants were built by using the GLYCAM online tool “Glycoprotein Builder”42. The “Glycoprotein Builder” was used to select the glycosylation site and attach the glycan to proteins to form either O- or N-linked glycoproteins. After obtaining the initial 3D model of a given glycosylated variant, an initial energy minimization of the glycoprotein was performed using a steepest descent algorithm43. Finally, the electrostatic potential surface of the glycoprotein was built using Glycan Reader44.
Data analysis and statistics
The data was expressed as means ± SD unless indicated otherwise and n indicates the number of cell samples or mice. One-way ANOVA and Tukey’s multiple comparisons test were used to compare values among groups (Prism; GraphPad). Statistical significance was considered as p value ≤ 0.05.
References
Go, A. S. et al. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127, 143–152, doi:10.1161/CIR.0b013e318282ab8f (2013).
Chen, C. H. et al. Effects of electronegative VLDL on endothelium damage in metabolic syndrome. Diabetes care 35, 648–653, doi:10.2337/dc11-1623 (2012).
Lee, H. C. et al. VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation. Int J Mol Sci 17, doi:10.3390/ijms17010134 (2016).
Schnabel, R. B. et al. Development of a risk score for atrial fibrillation (Framingham Heart Study): a community-based cohort study. Lancet 373, 739–745, doi:10.1016/S0140-6736(09)60443-8 (2009).
Aro, A. L. et al. Prognostic significance of prolonged PR interval in the general population. Eur Heart J 35, 123–129, doi:10.1093/eurheartj/eht176 (2014).
Doka, G., Radik, M., Krenek, P., Kyselovic, J. & Klimas, J. 1a.02: Microrna-208a and its Host Gene Cardiac Myosin Heavy Chain Myh6 are Involved in Hypertrophic Heart Dysfunction. J Hypertens 33(Suppl 1), e1, doi:10.1097/01.hjh.0000467352.59037.c9 (2015).
Magnani, J. W. et al. P Wave Indices, Obesity, and the Metabolic Syndrome: the Atherosclerosis Risk in Communities Study. Obesity (Silver Spring, Md.) 20, 666–672, doi:10.1038/oby.2011.53 (2012).
Cheng, S. et al. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA 301, 2571–2577, doi:10.1001/jama.2009.888 (2009).
Nielsen, J. B. et al. P-wave duration and the risk of atrial fibrillation: Results from the Copenhagen ECG Study. Heart Rhythm 12, 1887–1895, doi:10.1016/j.hrthm.2015.04.026 (2015).
Haugan, K. et al. Rotigaptide (ZP123) improves atrial conduction slowing in chronic volume overload-induced dilated atria. Basic & clinical pharmacology & toxicology 99, 71–79, doi:10.1111/j.1742-7843.2006.pto_432.x (2006).
Sawaya, S. E. et al. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292, H1561–1567, doi:10.1152/ajpheart.00285.2006 (2007).
Guerra, J. M., Everett, T. H. T., Lee, K. W., Wilson, E. & Olgin, J. E. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 114, 110–118, doi:10.1161/CIRCULATIONAHA.105.606251 (2006).
Ueda, N., Yamamoto, M., Honjo, H., Kodama, I. & Kamiya, K. The role of gap junctions in stretch-induced atrial fibrillation. Cardiovasc Res 104, 364–370, doi:10.1093/cvr/cvu202 (2014).
Polontchouk, L. et al. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J Am Coll Cardiol 38, 883–891 (2001).
Kirubakaran, S. et al. Fractionation of electrograms is caused by colocalized conduction block and connexin disorganization in the absence of fibrosis as AF becomes persistent in the goat model. Heart Rhythm 12, 397–408, doi:10.1016/j.hrthm.2014.10.027 (2015).
Shi, H. F. et al. Prevalence and spectrum of GJA5 mutations associated with lone atrial fibrillation. Molecular medicine reports 7, 767–774, doi:10.3892/mmr.2012.1252 (2013).
Johnstone, S. R., Billaud, M., Lohman, A. W., Taddeo, E. P. & Isakson, B. E. Posttranslational modifications in connexins and pannexins. J Membr Biol 245, 319–332, doi:10.1007/s00232-012-9453-3 (2012).
Issad, T. O-GlcNAcylation of connexin 40: a sweet connection between diabetes and endothelial cell dysfunction? Focus on “O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice”. American Journal of Physiology - Cell Physiology 309, C590–C592, doi:10.1152/ajpcell.00260.2015 (2015).
Makino, A. et al. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. American Journal of Physiology - Cell Physiology 309, C593–C599, doi:10.1152/ajpcell.00069.2015 (2015).
Kaese, S. & Verheule, S. Cardiac electrophysiology in mice: a matter of size. Frontiers in physiology 3, 345, doi:10.3389/fphys.2012.00345 (2012).
Tsai, W. C. et al. Small Conductance Calcium Activated Potassium Current and the Mechanism of Atrial Arrhythmia in Mice with Dysfunctional Melanocyte-like Cells. Heart rhythm: the official journal of the Heart Rhythm Society, doi:10.1016/j.hrthm.2016.03.011 (2016).
Simon, A. M., Goodenough, D. A. & Paul, D. L. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Current biology: CB 8, 295–298 (1998).
Leaf, D. E. et al. Connexin40 imparts conduction heterogeneity to atrial tissue. Circ Res 103, 1001–1008, doi:10.1161/circresaha.107.168997 (2008).
Foote, C. I., Zhou, L., Zhu, X. & Nicholson, B. J. The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. The Journal of cell biology 140, 1187–1197 (1998).
Mitchell, G. F., Jeron, A. & Koren, G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol 274, H747–751 (1998).
Luo, T. et al. Characterization of atrial histopathological and electrophysiological changes in a mouse model of aging. International journal of molecular medicine 31, 138–146, doi:10.3892/ijmm.2012.1174 (2013).
Takahashi, K. et al. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J Mol Cell Cardiol 90, 38–46, doi:10.1016/j.yjmcc.2015.11.034 (2016).
Lo, C. W. Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res 87, 346–348 (2000).
Chaldoupi, S. M., Loh, P., Hauer, R. N., de Bakker, J. M. & van Rijen, H. V. The role of connexin40 in atrial fibrillation. Cardiovasc Res 84, 15–23, doi:10.1093/cvr/cvp203 (2009).
Eckardt, D. et al. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J Mol Cell Cardiol 36, 101–110 (2004).
Kato, T., Iwasaki, Y.-k & Nattel, S. Connexins and Atrial Fibrillation: Filling in the Gaps. Circulation 125, 203–206, doi:10.1161/circulationaha.111.075432 (2012).
Laird, D. W. The life cycle of a connexin: gap junction formation, removal, and degradation. J Bioenerg Biomembr 28, 311–318 (1996).
Wang, J. et al. Lipoprotein subclass profiles in individuals with varying degrees of glucose tolerance: a population-based study of 9399 Finnish men. J Intern Med 272, 562–572, doi:10.1111/j.1365-2796.2012.02562.x (2012).
Alonso, A. et al. Blood lipids and the incidence of atrial fibrillation: the multi-ethnic study of atherosclerosis and the framingham heart study. J Am Heart Assoc 3, doi:10.1161/jaha.114.001211 (2014).
Fauchier, L., Clementy, N. & Babuty, D. Statin therapy and atrial fibrillation: systematic review and updated meta-analysis of published randomized controlled trials. Curr Opin Cardiol 28, 7–18, doi:10.1097/HCO.0b013e32835b0956 (2013).
Isik, T. et al. Relation of the metabolic syndrome with proarrhythmogenic electrocardiographic parameters in patients without overt diabetes. Acta Cardiol 67, 195–201, doi:10.2143/AC.67.2.2154210 (2012).
Hanci, V. et al. Preoperative abnormal P and QTc dispersion intervals in patients with metabolic syndrome. Anesth Analg 112, 824–827, doi:10.1213/ANE.0b013e3181f68ff8 (2011).
Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 285, 2486–2497 (2001).
Eswar, N. et al. Comparative protein structure modeling using Modeller. Current protocols in bioinformatics/editoral board, Andreas D. Baxevanis… [et al.] Chapter 5, Unit 5 6, doi:10.1002/0471250953.bi0506s15 (2006).
Maeda, S. et al. Structure of the connexin 26 gap junction channel at 3.5 angstrom resolution. Nature 458, 597–U561, doi:10.1038/nature07869 (2009).
Kao, H. J. et al. A two-layered machine learning method to identify protein O-GlcNAcylation sites with O-GlcNAc transferase substrate motifs. BMC bioinformatics 16(Suppl 18), S10, doi:10.1186/1471-2105-16-S18-S10 (2015).
Ceroni, A., Dell, A. & Haslam, S. M. The GlycanBuilder: a fast, intuitive and flexible software tool for building and displaying glycan structures. Source code for biology and medicine 2, 3, doi:10.1186/1751-0473-2-3 (2007).
Arfken, G. The Method of Steepest Descents in Mathematical Methods for Physicists. 3 edn (Orlando, 1985).
Jo, S., Song, K. C., Desaire, H., MacKerell, A. D. Jr. & Im, W. Glycan Reader: automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J Comput Chem 32, 3135–3141, doi:10.1002/jcc.21886 (2011).
Acknowledgements
The authors thank the technical support for confocal microscopic analysis of connexins from Dr. Yu-Jun Lai and Dr. Hung-I Yeh at the Heart and Circulatory Physiology Laboratory of Medical Research Department, Mackay Memorial Hospital, New Taipei City, Taiwan. We also thank the technical support for optical mapping system from Dr. Shien-Fong Lin at Institute of Biomedical Engineering, National Chiao Tung University, Hsinchu, Taiwan. We thank Liang-Yin Ke for technical assistance in VLDLs preparation. We thank I-Chieh Huang for his assistance in the manuscript revision. This study was funded by the Taiwan Ministry of Science and Technology (MOST 104-2314-B-037-080-MY3 to HC Lee), grants from Kaohsiung Medical University Hospital (KMUH103-3T03 and KMUH104-4T04 to HC Lee), and grants from Kaohsiung Medical University (KMU-TP103D05, KMU-TP104D04, and KMU-TP105D08 to HC Lee).
Author information
Authors and Affiliations
Contributions
H.C. Lee wrote the paper, the research projects, conceptualized the ideas and study design. C.C. Chen established the protein structural modeling platform and carried out the computational simulation. W.C. Tsai established the optical mapping setup, protocols, and carried out experiments. H.C. Lee, H.T. Lin, Y.L. Shiao performed the experiments and data analyses. B.N. Wu, S.H. Sheu, W.T. Lai, and C.H. Chen supervised the research activities, contributed analysis tools. W.T. Lai revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, HC., Chen, CC., Tsai, WC. et al. Very-Low-Density Lipoprotein of Metabolic Syndrome Modulates Gap Junctions and Slows Cardiac Conduction. Sci Rep 7, 12050 (2017). https://doi.org/10.1038/s41598-017-11416-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11416-5
This article is cited by
-
The roles of connexins and gap junctions in the progression of cancer
Cell Communication and Signaling (2023)
-
Association of lipid composition and unsaturated fatty acids of VLDL with atrial remodeling in metabolic syndrome
Scientific Reports (2023)
-
The role of postprandial very-low-density lipoprotein in the development of atrial remodeling in metabolic syndrome
Lipids in Health and Disease (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
