
Abstract
Cucumis melo is highly diverse for fruit traits providing wide breeding and genetic research opportunities, including genome-wide association (GWA) analysis. We used a collection of 177 accessions representing the two C. melo subspecies and 11 horticultural groups for detailed characterization of fruit traits variation and evaluation of the potential of GWA for trait mapping in melon. Through genotyping-by-sequencing, 23,931 informative SNPs were selected for genome-wide analyses. We found that linkage-disequilibrium decays at ~100 Kb in this collection and that population structure effect on association results varies between traits. We mapped several monogenic traits to narrow intervals overlapping with known causative genes, demonstrating the potential of diverse collections and GWA for mapping Mendelian traits to a candidate-gene level in melon. We further report on mapping of fruit shape quantitative trait loci (QTLs) and comparison with multiple previous QTL studies. Expansion of sample size and a more balanced representation of taxonomic groups might improve efficiency for simple traits dissection. But, as in other plant species, integrated linkage-association multi-allelic approaches are likely to produce better combination of statistical power, diversity capture and mapping resolution in melon. Our data can be utilized for selection of the most appropriate accessions for such approaches.
Similar content being viewed by others


Introduction
Melon (Cucumis melo L.: Cucurbitaceae) contains a wealth of phenotypic diversity for multiple attributes, especially for fruit traits such as size, shape, external (rind) and internal (flesh) color, sugar content, acidity, texture and aroma1. This wide diversity is the source for ongoing genetic research and breeding aimed at mapping and identifying quantitative trait loci (QTLs) and genes affecting key horticultural and consumers’ preference traits. Numerous genetic studies in recent years focused on fruit quality traits in melon, including fruit size and shape2,3,4,5, flesh color6,7,8,9, rind color10, netting and sutures11, 12, sweetness and aroma13, 14, acidity15 and ripening behavior16. Most of these studies utilized targeted bi-parental populations for the genetic analyses and trait mapping.
Bi-parental linkage mapping was, to a large extent, the default genetic mapping approach for simple and quantitative traits in plant species where generation time is short and the development of segregating populations is feasible. This approach is based on the analysis of the segregation of polymorphism between the parental lines and their progeny. Various linkage population types are commonly used in plant genetics, such as: F2:3, recombinant inbred lines (RILs), double-haploids (DH), introgression lines (ILs) or backcross inbred lines (BILs). While such populations display different attributes with respect to time and cost of creation, statistical power and genetic resolution, they all share the advantages and disadvantages of bi-allelic, non-structured designs.
Approximately 15 years ago, association mapping methodology started to become a valid alternative strategy for trait mapping in plants17, 18. Among the advantages of association genetics is the use of existing collections, the ability to simultaneously analyze wide phenotypic diversity resulting from multi-allelic genetic variation, and the exploitation of accumulated historical recombination events. A key limiting factor that inhibited the effective implementation of this approach in plant genetics was the inherent effect of population structure on creation of spurious associations. The development and successful application of statistical and computational tools to control for genetic relatedness19,20,21 enhanced the use of this approach in plants. At first, the implementation of association mapping was mostly through the analysis of candidate-genes, due to the insufficient genome-wide marker coverage defined by linkage-disequilibrium (LD) decay profile and genome size22,23,24. However, with the growing application of NGS-based approaches for high-density genotyping, the genome wide association study (GWAS) approach is now extensively used for genome-wide genetic dissection of traits in many crop plants25, 26.
Several recent studies in melon have used germplasm collections for characterization of diversity, population structure and for trait mapping. Tomason et al. screened 87 melon accessions with 286 simple sequence repeat (SSR) markers to describe population structure and fruit traits variation27. Leida et al. used a set of 175 melon accessions genotyped with 251 single nucleotide polymorphisms (SNPs; 148 background SNPs and 103 within candidate genes for sugar metabolism and ripening) to describe population structure, LD and candidate gene associations for sugar accumulation and ripening behavior28. Two recent studies used genotyping-by-sequencing (GBS) to genotype melon panels and to describe population structure and LD using genome-wide high density markers coverage (13,789 SNPs and 25,422 SNPs)29, 30.
In the current study a diverse collection of 177 melon accessions was extensively phenotyped for a number of traits and genotyped with 23,931 SNPs using GBS. The main objective was to describe genetic properties of this diversity panel and to test the actual potential and effectiveness of the GWAS approach for trait mapping in melon. Population structure and genome-wide LD patterns are described. Through GWA analysis, we demonstrate the mapping of several Mendelian traits to a narrow interval overlapping with known causative genes, providing a first proof-of-concept for the potential of association genetics for high resolution mapping to a candidate-gene level in melon.
Results
Phenotypic diversity across GWAS panel
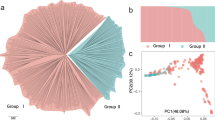
A diversity panel of 177 inbred melon accessions was used in the current study. This panel represents the two melon subspecies (melo and agrestis), encompassing 11 horticultural groups (Fig. 1A,B Table S1). The collection was grown in a replicated trial in the open field at Newe Ya’ar in summer 2015 and phenotyped for fruit traits during fruit development and at maturity. Images were taken on developing fruits of all accessions throughout the season from anthesis till harvest. Fifteen mature fruits per accession were harvested from three replicated plots for phenotyping which allowed us to obtain high heritability values for the measured traits. One of the key advantages of using diverse collections compared to bi-parental segregating populations is the extended range of phenotypic variation captured across multiple traits. Figure 2 shows normalized phenotypic ranges (ratio between min and max entry means) in three bi-parental melon populations, relative to the range measured in the GWAS panel, where the ratio is standardized to 1, across numerous selected phenotypic traits (Table S2). These reference experimental bi-parental populations are derived from wide crosses aimed at mapping key fruit quality traits (details at the materials and methods and Figure S1). Range for fruit weight, for example, in the GWAS panel is between 50 g for the smallest accession and 5,000 g for the largest (100-fold difference) while the maximum polymorphism for this trait in the bi-parental populations is 5-fold for the SAS × DOYA cross. For total soluble solids (TSS) the range across the GWAS panel is between 3.5% and 16.2% TSS (4.6-fold) while the ranges within the bi-parental populations are 3-, 2.4- and 1.1-fold. Flesh color is another example where the GWAS panel capture wide phenotypic spectrum in melon (from orange, through white, to green including the different intensities within each color category) and each of the bi-parental crosses captures only a fraction of this spectrum (SAS × DOYA: green, TAD × DUL: green-orange, PI × DUL: orange). The overall extended phenotypic variation captured within the GWAS panel compared to bi-parental populations is evident in our comparison also for time to maturity (i.e. earliness), fruit shape, rind stripes, ripening behavior and many of the other attributes that were measured.

Germplasm composition and structure. (A) Pie chart of the frequencies of the different horticultural groups across the melon collection. (B) Genetic PCA plot colored by horticultural group.

Comparison of normalized phenotypic variation between the GWAS collection and bi-parental melon populations for various traits. Phenotypic range is expressed as the ratio between maximum and minimum entry means for each set where all values are adjusted relative to the range at the GWAS panel (that is therefore normalized to 1).
Genetic variation and genome-wide LD patterns
The collection was genotyped using GBS approach31. Over 500,000,000 good barcoded reads were produced and 4,213,896 sequence-tags were extracted with minimum of 3 reads per tag. Fifty-five percent of the tags were uniquely aligned, 2.1% were aligned to more than one locus and 42.9% were unaligned. Such alignment rate for GBS tags in a plant genome can result from several possible factors: (1) Short tag sequence (too short to align significantly), (2) Tag sequence contains highly repetitive DNA, (3) The haplotype is too divergent from the reference to align, (4) Structural variation: the tag sequence is not present in the reference genome individual, or (5) Incomplete reference: the tag sequence is not present in the reference genome.
A total of 99,263 SNPs that were mapped to the melon genome32 were identified across the 177 melon accessions. Following further filtration to minor allele frequency (MAF) > 0.05 and maximum of 6% missing data per site, 23,931 informative SNP markers were defined as the genotypic dataset in this study. As expected in GBS, SNP densities fluctuate within and between chromosomes, resulting in differential coverage across the genome (Figure S2) with a genome-wide average density of ~1 SNP/18 Kb. Principal component analysis (PCA) was performed using the whole-genome marker data. Two-dimensional PCA plot using PCA component#1 and PCA component#2 is presented in Fig. 1B where the 11 horticultural groups are color-coded. In general, good consensus is observed between the phenotypically defined groups and the clustering based on genetic relatedness, with an obvious separation between the melo and agrestis sub-species (also shown as a phylogenetic tree in Figure S3). Also, the inodorous group is nicely clustered and separated from the reticulatus and cantalupensis types that cluster together with wide overlapping genetic variation within them. An admixture-based clustering model implemented in the software STRUCTURE33 was also used to infer the genetic structure of the collection. Clear division between the two subspecies was obtained at K = 2 across the whole collection, and K = 7 provided the second best fit for sub-grouping the accessions, in agreement with the taxonomic classification (Figure S4). We then used the genome-wide marker set to characterize patterns of LD across the genome. LD was calculated as R2 between SNP pairs and plotted against their physical distance. On a genome-wide average, LD decay in this melon collection within ~100 Kb to a level below R2 = 0.2 (Fig. 3). Substantial variation in local LD patterns that exists along and between chromosomes is shown in Figures S2 and S5, where LD is plotted against physical positions or distance. Based on the genome-wide LD decay pattern (~100 Kb) and melon genome size (~450 Mb), the estimated minimal number of markers required for efficient genome-wide scan using a panel similar in composition to the one used here would be ~5,000.

Genome-wide LD decay plot. R2 between intra-chromosomal marker pairs plotted against the physical distance between them.
Whole genome LD mapping
While LD decay analysis provides a general averaged view on potential genetic resolution, we took a direct comparative mapping approach. To test the usefulness of GWAS approach for mapping traits using the melon diversity collection, we phenotyped and performed association mapping for several high heritability simple traits where causative genes are known. The obtained mapping results provide insight into local resolutions that can be achieved using this platform.
Characterization and mapping of sex determination trait
In melon, most plants are either monoecious or andromonoecious. Monoecious female flowers contain only the female reproductive organs (carpels) and andromonoecious contain both female and male (stamens) organs. This trait was previously mapped to the a locus34 on chromosome 2 and the causative gene (CmACS-7) was subsequently cloned35. In the current study, female flowers at anthesis were visually characterized for their sex-expression type (monoecious or andromonoecious, Fig. 4a) across all accessions. Sixteen percent of the accessions in the collection are monoecious and 84% are andromonoecious. Projection of the sex expression phenotypes on the genetic PCA plot provides an informative view on the distribution of the phenotypic classes across the genetic variation and population structure (Fig. 4b). While the main C. agrestis cluster in our sample (containing makuwa, chinensis and conomon accessions) is monomorphic, showing only the andromonoecious phenotype, for the rest of our panel the distribution of the phenotypic classes is relatively uniform across the genetic variation and independent of population structure, which is an important attribute for effective association analysis. Using these phenotypes we then performed GWA analysis and found a single significant locus on chromosome 2 associated with this trait (P = 1.4 × 10−12 at mixed linear model (MLM) analysis, Fig. 4c,d). Zooming in on the genomic neighborhood of this locus showed that the peak of the trait locus is at position 1,771,409, ~60 Kb and one gene apart from the CmACS-7 gene, in which we did not have a SNP in our genotyping set (MELO3C015444: Chr2 1,708,995–1,711,002, Fig. 4e). To further validate the peak marker SNP for this trait (SNP1771409), we have genotyped a set of lines from the panel using PCR marker at the CmACS-7 gene sequence35. High LD is found between SNP1771409 and the CmACS-7 PCR marker (Figure S6). Figure 4e also shows the decline of significance bellow the genome-wide threshold at a less than 100 KB window around the trait-locus peak, supported by the local LD pattern around this locus (Figure S7).

Mapping of flower sex-expression. (a) Examples of monoecious (right) and andromonoecious (left) female flowers at anthesis in the diversity panel. (b) Female flower types projected on genetic PCA plot. (c) Manhattan plot of GWA of female flower type. Associations were tested using mixed-linear model (MLM_Q + K) for controlling population structure and relatedness. Genome-wide significance threshold is adjusted for multiple comparisons at FDR5%. (d) Quantile-quantile (Q-Q) plot for distribution of P values at the MLM_Q + K model. The negative logarithm of the observed (y axis) and the expected (x axis) P value is plotted for each SNP (dot). The gray dashed line indicates the null hypothesis. (e) Zoom in on chromosome 2 peak where causative gene CmACS-7 35 (MELO3C015444) is shown.
Characterization and mapping of flesh color
Three major flesh color categories are defined in melon; green, white and orange, with β-carotene and chlorophyll being the predominant pigments of the orange and green phenotypes, respectively. The major locus differentiating between orange and non-orange flesh is gf, previously mapped to chromosome 97. Recently, the gene CmOr was identified as gf and accounts for most of the orange/non-orange variation in melon9. In order to characterize flesh color variation in the GWAS panel, longitudinal sections of fifteen fruits per accession were scanned and analyzed for multiple characteristics, including flesh color, using the Tomato Analyzer software36 (Fig. 5a). Nine of the fruits from each accession were also measured for flesh color using handheld colorimeter. Image-based color values were highly correlated with colorimeter measurements across 1500 fruits that were measured in parallel (R = 0.94, Figure S8). Overall flesh color showed very high broad-sense heritability in our experiment (H2 = 0.96, Figures S9 and S10). Flesh color phenotypes distribute uniformly within the GWAS panel with all three color categories represented in a significant proportion (Fig. 5b). Projection of flesh color variation on the genetic PCA plot reveals the strong relationship between this trait and population structure (Fig. 5c). A large proportion of the orange accessions class in the reticulatus group and most of the white flesh accessions are in the inodorous group. Agrestis accessions are mostly white flesh. This non-random distribution and clear dependence between the genetic landscape and phenotypic variation is expected to yield excessive spurious associations if a simple statistical model is used for association analysis. This confounding effect can be controlled through the use of relatedness estimates as cofactor in MLM analysis21. We performed GWA analysis using both statistical models. As expected, the generalized linear model (GLM) resulted in high proportion of significant SNP effects across all chromosomes (Fig. 5d). However, a single SNP on chromosome 9 (at position 20,550,439) was noticeable for its strong and highly significant effect (P = 10−34). MLM analysis removed most of the significant effects (a mix of mostly spurious and maybe some positive associations) and only the chromosome 9 SNP remained highly significant (P = 3.7 × 10−12) and passed the genome-wide threshold (false discovery rate (FDR) 5%, Fig. 5e,f). Zooming in on this SNP reveals that it is located within the causative CmOr gene (MELO3C0005449: Chr9 20,548,319–20,555,636). This SNP has MAF of 0.32, it explain ~70% of the flesh color variation across the panel and effectively distinguish between orange and non-orange (Fig. 5g). The closest SNPs around this trait-peak marker are 40 Kb apart. This is a cluster of 3 SNPs in LD with each other that show P values of ~10−4, indicating the decline of LD at less than 100 Kb in this region. To further validate SNP20550439, we have genotyped all the lines in the panel for the polymorphism in the CmOr gene, as described by Tzuri et al. (2015)9, and obtained 99% match in allelic segregation (Figure S11). In consensus with the local LD decay in this region (Figures S2 and S12), this example also demonstrates the decline of significance within a relatively short interval around the peak, producing a manageable window of candidates that include the causative gene.

Mapping of flesh color. (a) Example of scanned longitudinal sections of melons from the diversity panel. 2,650 fruits were scanned and analyzed using Tomato Analyzer software36. (b) Frequency distribution of entry mean hue index as measured from fruit scans. Bar colors reflect the visual perception. (c) Flesh hue indexes projected on genetic PCA plot. (d) Manhattan plot of GWA of flesh color. Associations were tested using naïve General-linear model (GLM) (e) Manhattan plot of GWA of flesh color. Associations were tested using mixed-linear model (MLM_Q + K) for controlling population structure and relatedness. Genome-wide significance threshold is adjusted for multiple comparisons at FDR5%. (f) Quantile-quantile (Q-Q) plot for distribution of P values at the MLM_Q + K model. The negative logarithm of the observed (y axis) and the expected (x axis) P value is plotted for each SNP (dot). The gray dashed line indicates the null hypothesis. (g) Allelic effect plot for the SNP within CmOr gene (MELO3C005449). Each point represents accession mean color (hue index) and points are color-coded based on the visual color perception.
Characterization and mapping of yellow rind color
Melon accessions with yellow rind are present in our GWAS panel. We previously reported that the yellow color is caused by the accumulation of naringenin chalcone, a yellow flavonoid pigment37. A Kelch domain-containing F-box protein coding gene (CmKFB) on chromosome 10 was identified as causative for the yellow rind phenotype in yellow casaba melon accession (C. melo, var inodorus)10. We characterized the collection for rind color through visual scoring and selected only smooth rind (without netting that could mask the rind color) accessions that are either yellow or white/cream (Fig. 6a) for association analysis. Sixty-nine accessions remained and were included in the yellow rind association analysis, 37 accessions with white/cream rind and 32 with yellow rind. Projection of this trait on the genetic PCA plot is showing population structure dependence for the phenotypic distribution with a high proportion of the white rind in the reticulatus group and most yellow accessions in inodorous. However, both agrestis and melo sub-species show polymorphism for this trait (Fig. 6b). GWAS was performed using both GLM and MLM approaches and the effect of population structure control on genome-wide significance levels are evident (Fig. 6c,d). While sample size used for this analysis is fairly small (due to the exclusion of netted and orange or green rind accessions), the strongest genome-wide effect that we found for this trait, on chromosome 10, was still marginally significant at FDR30% (P = 4 × 10−5, MLM). Zooming in on this region showed two SNPs at positions 3,541,676 and 3,541,866 (190 bp apart) that are in complete LD (R2 = 1). These SNPs, that showed the strongest association with yellow rind trait, are located 65 Kb and 8 genes away from the CmKFB gene10 (MELO3C011980: Chr10 3,475,253–3,476,386, Fig. 6e). SNP3541676 has MAF of 0.45 across the ‘rind color’ panel and it is associated with rind color in 90% of the accessions (Fig. 6f). To further validate SNP3541676 and SNP3541866 at the QTL peak, we genotyped the yellow and white rind accessions using PCR marker at the CmKFB gene10 and good co-segregation with the peak markers was shown (Figure S13). Local LD decline pattern for this genomic region fits with the genome-wide average and supports the obtained resolution (Figures S2 and S14). It is important to note that yellow rind phenotyping was based here only on visual observation and not supported by chemical analysis of flavonoids or carotenoids. Therefore, it could be that some of the fruits defined as yellow in fact accumulate carotenoids rather than naringenin chalcone. Moreover, melons also exhibit genetic variability for waxiness of the cuticle38, 39. A waxy epicuticular layer can mask rind colors and lead to modified visual phenotyping. These factors may have introduced some ‘noise’ to our mapping, further reducing the significance of the Chr10 QTL.

Mapping of yellow rind trait. (a) Examples of some fruits with yellow and white rinds used for the analysis of the trait. (b) Yellow or white rind colors projected on genetic PCA plot. (c) Manhattan plot of GWA of rind color (both simple model (GLM) and population structure corrected model (MLM_Q + K) are presented). (d) Quantile-quantile (Q-Q) plot for distribution of P values at the MLM_Q + K model. The negative logarithm of the observed (y axis) and the expected (x axis) P value is plotted for each SNP (dot). The gray dashed line indicates the null hypothesis. (e) Zoom in on chromosome 10 peak where causative gene CmKFB (MELO3C011980)10 is shown. (f) Contingency analysis for distribution of rind color across alleles at Chr10_SNP3541676.
Characterization and mapping of fruit shape
Extensive diversity is present in C. melo for fruit size and shape. Fruit weight varies 100-fold within our collection (50 g–5000 g) and fruit shape index (ratio between length and width) varies between 0.66 and 6.3 (10-fold) across the panel (Fig. 7a,b, Table S2, Figure S15). Fruit shape is an important trait from breeding perspective as, alongside other fruit attributes such as rind netting, flesh color and ripening behavior, it defines horticultural and marketing groups. While the components of fruit shape (fruit length and width) may be influenced by environmental conditions (as they reflect growth rate), shape index seem to be a very coordinated and conserved attribute. Furthermore, fruit shape is programmed early on in fruit development process as there is good correlation between ovary and mature fruit shape indexes in melon40, 41. The collection was characterized for fruit shape through image-analysis-based measurements taken on digital scans of longitudinal fruit sections (Fig. 7a, see materials and methods). Fifteen fruits per accession were measured and the average shape index was calculated. The extensive diversity and wide sampling resulted in very high heritability obtained for this trait (H2 = 0.95, Figure S15). Projection of fruit shape means on the genetic PCA revealed the distribution of shape variation across the genetic landscape and the effect of population structure on phenotypic distribution (Fig. 7c). Most of the elongated fruits (index > 1.5) belong to C. melo ssp. melo var. inodorous and flexuosus, and C. melo ssp. agrestis var. makua. Next, we mapped fruit shape through GWA analysis. Significant SNPs in MLM analysis (at FDR5%) were found on all chromosomes (Fig. 7d, Table S3). We then compared our significant SNPs with previously mapped fruit shape QTLs from multiple studies that have been summarized into a consensus map5, 12. The polygenic architecture of fruit shape observed here is in agreement with these previous results where fruit shape QTLs were mapped in different studies to eleven out of the twelve melon chromosomes. Monforte et al.5 identified meta-QTLs for fruit shape on five chromosomes where common intervals were identified in several studies. We found significant SNPs (at Bonferroni adjusted threshold) on four of these chromosomes (FSQM-2, 8, 11 and 12, Fig. 7d,e and Table S3). On chromosomes 3, 6, and 10 we found significant SNPs that overlapped with QTLs that were reported in a previous study (FSQC3.5, FSQC6.4, FSQX6.1, FSQA10.1, FSQC10.25). Interestingly, large effects appear in higher frequencies for SNPs with MAF < 0.1 (Figure S16), while the distribution of allele frequencies is relatively uniform across our genotyping set filtered to MAF > 0.05 (Figure S17). Similar patterns of negative correlations between polymorphism effect size and allele frequency were also shown for multiple traits in maize42, 43.

Characterization and mapping of fruit shape. (a) Examples of elongated and round fruits from the collection and the measurements taken for shape analysis. (b) Frequency distribution of fruit shape index for accession means. (c) Fruit shape index projected on genetic PCA plot. (d) Manhattan plots of GWA of fruit shape index. Upper panel: GLM analysis. Lower panel: MLM_Q + K analysis. Dashed gray horizontal lines are genome-wide significance thresholds adjusted for multiple comparisons using FDR5% and Bonferroni corrections. Horizontal bars bellow the plot indicate names and positions of previously mapped QTLs5. Dark red are meta-QTLs. Pink are QTLs identified in one or two experiments. (e) Quantile-quantile (Q-Q) plot for distribution of P values at the MLM_Q + K model. The negative logarithm of the observed (y axis) and the expected (x axis) P value is plotted for each SNP (dot). The gray dashed line indicates the null hypothesis.
Discussion
The power of combining NGS-based genotyping and GWAS for achieving mapping to the gene level resolution was demonstrated in several recent studies in plants44, 45. The ability to simultaneously screen diverse germplasm collections and map traits through GWAS was described as a mean to improve tomato flavor46, or identify candidate-genes for manganese efficiency in barely47.
The current study is aimed at establishing melon diversity collection as a permanent germplasm resource for discovery of phenotypic variation and genetic mapping of traits. We performed phenotypic and genotypic analyses to test and demonstrate the genetic resolution and overall effectiveness of this platform. The panel used in this study is composed of 177 accessions that represent large portion of the genetic and phenotypic diversity within C. melo. The collection was genotyped with 23,931 SNPs that cover all 12 melon chromosomes at an average density of ~1 SNP per 18 Kb. This density and coverage allowed us to calculate the extent of LD and show that LD decays on average across ~100 KB in this panel (Fig. 3), defining the potential mapping resolution and estimated number of markers required for GWAS in this crop. Since LD decay pattern reflects an average genetic attribute that describes theoretical mapping resolution, we performed real mapping of traits to test the actual resolution obtained. Three traits with high heritability, simple genetic basis and known causative genes were used as test cases: Sex-expression trait was mapped to chromosome 2, one gene away from the previously identified causative CmACS-7 gene35. Major flesh color locus was mapped within the causative CmOr gene that was mapped earlier using bi-parental linkage analyses7, 34 and later cloned using candidate gene approach9. The yellow rind trait locus was mapped 65 KB from the causative CmKFB gene that was previously identified through RNA-Seq bulk-segregant analysis (BSA) on F3s10.
Thus, three independent single gene traits were successfully mapped in the current study to narrow genomic intervals, where the most significant SNPs identified for each trait were located within less than 100 Kb windows from the known, previously mapped, causative genes. LD around these mapped trait loci decayed within the expected physical distance calculated at the whole genome level and allowed us to define confidence intervals containing less than 20 annotated open-reading frames (ORFs) in these three examples.
Fruit shape is highly polymorphic trait that has been extensively studied in melon and shown to be governed by multiple QTLs2,3,4,5. We analyzed fruit shape variation across the diversity panel and mapped QTLs for this trait. The extended variation and high heritability enabled the identification of multiple significant SNPs across all chromosomes (Fig. 7d, Table S3). Strong signals were detected in chromosomes 2, 8 and 11 where meta-QTLs were previously described5, as well as significant effects in other genomic regions mapped in specific studies. Availability of a reference genome for melon32 facilitates effective comparative mapping through alignment of results from different studies into a common framework48. The added value of using multi-allelic population is demonstrated here for fruit shape through the simultaneous identification of QTLs that were previously found in different populations.
Visual projection of phenotypic variation on the genetic landscape (expressed as 2D PCA plots) is shown here as a useful instrument to evaluate the relation between population structure and trait variation and to gain perspective about the history of allelic variation in target traits. For example, one can hypothesize that the CmOr gene dominant mutation, which governs the accumulation of carotenoids (mainly β-carotene) and orange flesh color9, most likely occurred during sub-speciation as the orange allele is present only in one accession of the ssp. agrestis group in our collection (6%) while the frequency of the Orange allele at the ssp. melo group is 41%. The genotypic data localize the light-orange-flesh ssp. agrestis accession (PI414723) intermediate between the two sub-species, supporting this hypothesis (Fig. 5c). A second flesh color variant, wf, that is associated with the transition from white to green flesh and was previously described and mapped to chromosome 83, 49 is evident only within the ssp. melo group. The effect of breeding selection is reflected by the clear division between color categories within C. melo ssp. melo where most orange accessions belong to the reticulatus and cantalupensis groups and most white accessions are among inodorous. This trait is now readily transferred across genetic backgrounds using marker-assisted selection and it is expected that distribution of flesh color across horticultural groups will be more uniform with the development and distribution of new product combinations.
Population structure and cryptic relatedness may lead to false-positive discoveries in association analyses50. The correlation between population structure and trait distribution is defining the expected rate of spurious associations and the impact of correcting for structure and relatedness on distribution of P values21. We show in the current study the varying effect of population structure on GWA results across different traits in melon. The most striking example is ovary hairiness, spreading versus appressed, which determined subdivision of C. melo into two subspecies, ssp. melo (spreading) and ssp. agrestis (appressed)51. As expected, hairiness phenotypic distribution is completely correlated with sub-specific division and therefore excess of significant effects (false positive associations) are found genome-wide (Figures S18). Another trait where distribution of phenotypes is confounded with population structure is flesh color (Fig. 5c). The prominent difference in distribution of GWA P values between GLM and MLM analyses in both of these traits (Fig. 5d,e, Figure S18D,E, Figure S19) is a clear evidence for that. While the control for population structure reduced the false-discovery ‘noise’ but did not exclude the detection and mapping accuracy for the major flesh color gene CmOr, it has most likely eliminated other true-positive associations that explain quantitative variation in color6, 7, 11. Flesh color and ovary hairiness are therefore examples for traits where the confounding effect of population structure reduce the power of association mapping and highlight the advantage of non-structured linkage designs. Similar situation was described for flowering time and other traits in maize52. This limitation of association mapping approach has led to the development and implementation of alternative multi-allelic designs in plant53 and animal54 genetics that combine association and linkage properties. In maize, Nested-Association Mapping (NAM) and Multi-parent Advanced Generation InterCrosses (MAGIC) designs were demonstrated as efficient platforms for high resolution trait dissection55, 56. MAGIC design was also recently used in tomato to detect candidate SNPs underlying QTLs57.
Advances in genotyping technologies, computational and statistical tools are supporting the implementation of multi-allelic designs. The ability to simultaneously capture a wide spectrum of allelic variation for genetic mapping is appealing compared to traditional bi-parental designs, particularly in cases where multiple traits are targeted in parallel. The goal of the current study was to evaluate the potential of using diverse collection for LD mapping in Cucumis melo. The results presented here provide demonstration for the effectiveness of the melon diversity collection and GWAS approach to map simple traits to candidate-gene level. Based on experience and lessons from other crops, the next logical step to promote the efficient dissection of complex traits in melon would be the development of multi-allelic segregating populations that will overcome the inherent limitations of GWAS and can serve as a community resource for broader comparative genetics within the Cucurbitaceae. The genotypic and phenotypic infrastructure laid in this study can support the selection of balanced representative core panel for that purpose as shown for our current 25 founders core panel (Fig. 8, materials and methods), where for both phenotypic and genotypic plots, selected accessions are distributed uniformly and capture the diversity spectrum.

Core-panel (25 accessions) that represent genotypic and phenotypic spectrums across the diverse collection. (a) Two-dimensional genetic PCA plot. Core-panel accessions are shown in red. (b) Frequency distributions of various phenotypic traits. Core-panel accessions are highlighted in red.
Materials and Methods
Plant materials and field experiment
The Newe-Ya’ar melon diversity collection used in this study is comprised of 177 inbred accessions derived from many countries and encompassing the two melon subspecies (ssp. agrestis and ssp. melo) and 11 horticultural groups (Fig. 1, Table S1, Figures S3 and S4). Seeds of each of the accessions were sown in seedling trays in early March, 2015. Seedlings were transplanted in early April to the field at Newe Ya’ar (32°43′05.4″N 35°10′47.7″E). The seedlings were spaced 50 cm apart, on raised beds covered with silver-colored plastic mulch (Ginnegar), 200 cm between bed centers. Soil type was grumusol, and the plants were drip-irrigated and drip-fertilized, to approximately 180 L/m2 over the course of the growing season. Each accession was represented by three plots of five plants each; in a randomized block design (RCBD).
The bi-parental populations that were used as reference for phenotypic variation spectrum in this study are created with lines that are part of the diverse collection and are shown in Figure S1. The three populations are: (1) SAS × DOYA population (120 F4 lines); cross between a sweet round-fruited accession (C. melo ssp. agrestis, var makuwa) and the non-sweet elongated pickling melon (C. melo ssp. melo, var flexuosus). (2) TAD × DUL population (166 RILs; F7); cross between an honey-dew line (C. melo ssp. melo, var inodorus) and an American cantaloupe melon (C. melo ssp. melo, var reticulatus). (3) PI414723 x DUL population11 (99 RILs); a wide cross between a C. melo ssp. agrestis, var momordica accession and an American cantaloupe melon (C. melo ssp. melo, var reticulatus). These populations were grown during several years in the same location (Newe-Yaar) and similar growing regime.
Core panel of 25 founder lines for future multi-allelic segregating populations was selected based on multiple criteria derived from this study: initial tentative set (n = 40) was constructed to represent all the different horticultural groups in the diverse collection (based on traditional classification). Phenotypic profiles were then used as the second primary factor; the preliminary core set was projected on the distribution of the different traits to ensure phenotypic spectrum is well captured in the core panel (as illustrated in Fig. 8). Following required adjustments and narrowing of the set to n = 30, based on the first two steps, final set was selected to meet the 25 accessions target, taking into account maximum polymorphism information content (PIC) value and uniform distribution on genetic diversity plots (PCA and phylogenetic tree).
Phenotyping
The collection was subjected to phenotypic characterization of fruits throughout the growing season to reflect variation in fruit development, with most effort on mature fruits. Phenotyping during fruit development was largely based on images taken at the field using digital camera. Representative female flowers and fruits from each accession were pictured once a week from anthesis till harvest. Images were tagged and used for manual annotations of fruit traits. Melons were harvested when fully ripe, as defined by number of days after anthesis, rind color and firmness in the non-climacteric accessions and by abscission of the fruit from the peduncle (fruit stem) in climacteric accessions. Five mature fruits per plot were sampled in multiple harvests and total of 15 fruits per accession were analyzed. Due to large variation in maturity time across the diverse collection, the field was walked through on a two-daily basis from middle of June till end of July for selective harvest of mature fruits. Melons were weighed and cut longitudinally for external and internal imaging. Internal side of all fruits was scanned using a standard document scanner (Canon, Lide120). Three fruits per plot were used for flesh color measurement (three reads per fruit) using hand colorimeter (Minolta Sensing Inc, Minolta Chroma Meter Model CR-400, Osaka, Japan). Flesh and rind samples were taken as bulks from at least three fruits per plot and immediately frozen in 50 ml tubes in liquid nitrogen, for further biochemical and molecular analyses. Fruit internal scanned images were analyzed using the Tomato-Analyzer software36 for the extraction of color, shape and size attributes.
SNP genotyping
DNA isolation for GBS
Total genomic DNA extractions were performed on the177 accessions. DNA isolation was performed using the GenEluteTM Plant Genomic DNA miniprep kit (Sigma, St. Louis, MO). The quality of the DNA was analyzed by ND-1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE) and by electrophoresis on agarose gel. The concentration of DNA was estimated using Qubit® 2.0 Fluorometer (Life Technologies, Singapore) and Qubit® dsDNA BR Assay Kit (Life Technologies, Eugene, OR).
GBS analysis
DNA was shipped to the Institute for Genomic Diversity facility at Cornell University for GBS. GBS 96-plex libraries were prepared using the restriction enzyme ApeKI, following an established protocol31. Fragments were sequenced on an Illumina HiSeq. 2500 as 100 bp, single-end reads and aligned to the reference genome of C. melo 32 available at https://melonomics.net/files/Genome/Melon_genome_v3.5.1/. TASSEL pipeline v3.0.173 was used for sequence alignment and single nucleotide polymorphism (SNP) calling58. Further filtration was performed using TASSEL v5.2.3359; SNP list was filtered to MAF > 0.05 and maximum of 6% missing data per site.
Validation of trait peak markers
Association results for flower sex-expression, flesh color and yellow rind were validated by genotyping polymorphisms at the causative genes: polymorphism at the CmACS-7 gene (MELO3C015444) was genotyped using CAPs marker adapted from Boualem et al.35. A 234 bp amplicon was produced (Forward primer: AGATTCGCCGTATTTTGCTG, Reverse primer: CCCTCACAATTTTCCTCCAA), cleaved with Alu I restriction enzyme and seperated on a 2% agarose gel. For CmOr gene (MELO3C05449) polymorphism was genotyped based on the SNP described by Tzuri et al.9. 500 bp amplicons (Forward: CTCCTTGGTTTTCTTCATG, Reverse: CAACAAAACCCATCAAGTC) were sequenced and aligned across all samples. SNP at position 160 was called. Polymorphism at the CmKFB gene was genotyped using a protocol adapted from Feder et al.10. A 81 bp product was amplified (Forward: AACACTCAAAATTCACTAAATGGTCT, Reverse: TGTCGTAATTTTAATTTTACTTATTTTTATC) and a 23 bp indel differentiating between the alleles was scored on a 2.5% agarose gel.
Data analysis
Population structure, Kinship and LD analysis
TASSEL software (V 5.2.33)59 was used to estimate the relatedness matrix of pairwise kinship (k matrix) from the filtered SNP dataset using the Centered_IBS method60. Intra-chromosomal LD between pairs of sites was calculated in TASSEL on SNP set filtered to MAF > 0.15 to ensure reliable estimates. Phylogenetic tree was built using the neighbor-joining function in MEGA61. An admixture-based clustering model implemented in the software STRUCTURE33 was used to infer the genetic structure of the collection. Ten independent runs for each K value ranging from 1 to 10 were performed with a burn-in length of 100,000 and 100,000 Markov chain Monte Carlo (MCMC) repeats after burn-in. The optimal subpopulations number was calculated from the second order rate of change of likelihood (delta K method)62.
GWA mapping
In this study, four models (run in TASSEL) were used for the association analysis: the first used a generalized linear model (GLM) without any consideration for population structure; the second was GLM + Q where inferred ancestry of individuals (Q matrix) is used as covariate in the model; the third model was mixed linear model (MLM) using kinship matrix (k; random effect based on the genetic relatedness across all accessions) and the fourth model was MLM using both population structure (Q matrix) and relatedness (kinshp matrix).This model considered population structure and cryptic relationships thereby minimizing false positives and increasing the statistical power. Supplementary Figure S19 is showing Quantile-quantile (Q-Q) plots where distributions of P values at the different models are compared to the expected null hypothesis distribution. Significance threshold was corrected for multiple comparisons using the FDR approach63. For fruit shape GWA analysis, stringent Bonferroni correction was used to control for multiple comparisons.
References
Burger, Y. et al. Genetic variability for valuable fruit quality traits in Cucumis melo. Isr. J. Plant Sci. 54, 233–242 (2006).
Díaz, A. et al. Mapping and Introgression of QTL Involved in Fruit Shape Transgressive Segregation into ‘Piel de Sapo’ Melon (Cucucumis melo L.). PLoS One 9, e104188 (2014).
Monforte, A. J. et al. Identification of quantitative trait loci involved in fruit quality traits in melon (Cucumis melo L.). Theor. Appl. Genet. 108, 750–758 (2004).
Fernandez-Silva, I. et al. Shaping melons: Agronomic and genetic characterization of QTLs that modify melon fruit morphology. Theor. Appl. Genet. 121, 931–940 (2010).
Monforte, A. J., Diaz, A., Caño-Delgado, A. & Van Der Knaap, E. The genetic basis of fruit morphology in horticultural crops: Lessons from tomato and melon. J. Exp. Bot. 65, 4625–4637 (2014).
Cuevas, H. E., Staub, J. E., Simon, P., Zalapa, J. E. & McCreight, J. D. Mapping of genetic loci that regulate quantity of beta-carotene in fruit of US Western Shipping melon (Cucumis melo L.). Theor. Appl. Genet. 117, 1345–1359 (2008).
Cuevas, H. E., Staub, J. E., Simon, P. W. & Zalapa, J. E. A consensus linkage map identifies genomic regions controlling fruit maturity and beta-carotene-associated flesh color in melon (Cucumis melo L.). Theor. Appl. Genet. 119, 741–756 (2009).
Chayut, N. et al. Distinct mechanisms of the ORANGE protein in controlling carotenoid Flux. Plant Physiol. 173, 376–389 (2017).
Tzuri, G. et al. A ‘golden’ SNP in CmOr governs fruit flesh color of melon (Cucumis melo). Plant J. 82, 267–279 (2015).
Feder, A. et al. A Kelch domain-containing F-box coding gene negatively regulates flavonoid accumulation in Cucumis melo L. Plant Physiol. 169, 1714–1726 (2015).
Harel-Beja, R. et al. A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor. Appl. Genet. 121, 511–533 (2010).
Diaz, A. et al. A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol. 11, 111 (2011).
Zhang, H. et al. Mapping the flavor contributing traits on ‘Fengwei Melon’ (Cucumis melo L.) chromosomes using parent resequencing and super bulked-segregant analysis. PLoS One 11, e0148150 (2016).
Freilich, S. et al. Systems approach for exploring the intricate associations between sweetness, color and aroma in melon fruits. BMC Plant Biol. 15 (2015).
Cohen, S. et al. The PH gene determines fruit acidity and contributes to the evolution of sweet melons. Nat. Commun. 5, 4026 (2014).
Saladié, M. et al. Comparative transcriptional profiling analysis of developing melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genomics 16, 440 (2015).
Remington, D. L. et al. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA. 98, 11479–11484 (2001).
Thornsberry, J. M. et al. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28, 286–289 (2001).
Zhang, Z., Buckler, E. S., Casstevens, T. M. & Bradbury, P. J. Software engineering the mixed model for genome-wide association studies on large samples. Brief. Bioinform. 10, 664–675 (2009).
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360 (2010).
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–8 (2006).
Harjes, C. E. et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 319, 330–333 (2008).
Ehrenreich, I. M. et al. Candidate gene association mapping of arabidopsis flowering time. Genetics 183, 325–335 (2009).
Wilson, L. M. et al. Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16, 2719–2733 (2004).
Li, H. et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50 (2012).
Han, B. & Huang, X. Sequencing-based genome-wide association study in rice. Curr. Opin. Plant Biol. 16, 133–138 (2013).
Tomason, Y., Nimmakayala, P., Levi, A. & Reddy, U. K. Map-based molecular diversity, linkage disequilibrium and association mapping of fruit traits in melon. Mol. Breed. 31, 829–841 (2013).
Leida, C. et al. Variability of candidate genes, genetic structure and association with sugar accumulation and climacteric behavior in a broad germplasm collection of melon (Cucumis melo L.). BMC Genet. 16, 1–17 (2015).
Pavan, S. et al. Genotyping-by-sequencing of a melon (Cucumis melo L.) germplasm collection from a secondary center of diversity highlights patterns of genetic variation and genomic features of different gene pools. BMC Genomics 18, 59 (2017).
Nimmakayala, P. et al. Genome-wide differentiation of various melon horticultural groups for use in GWAS for fruit firmness and construction of a high resolution genetic map. Front. Plant Sci 7 (2016).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, 1–10 (2011).
Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. 109, 11872–11877 (2012).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Périn, C. et al. A reference map of Cucumis melo based on two recombinant inbred line populations. Theor. Appl. Genet. 104, 1017–1034 (2002).
Boualem, A. et al. A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science. 321, 836–838 (2008).
Rodríguez, G. R. et al. Tomato Analyzer: a useful software application to collect accurate and detailed morphological and colorimetric data from two-dimensional objects. JoVE, doi:10.3791/1856 (2010).
Tadmor, Y. et al. Genetics of flavonoid, carotenoid, and chlorophyll pigments in melon fruit rinds. J. Agric. Food Chem. 58, 10722–8 (2010).
Kubicki, B. Inheritance of some characters in muskmelons (Cucumis melo). Genet. Pol. 3, 265–274 (1962).
Dogimont, C. 2011 Gene List for Melon. Cucurbit Genet. Coop. Rep. 33–34 133, 104–133 (2011).
Eduardo, I. et al. Estimating the Genetic Architecture of Fruit Quality Traits in Melon Using a Genomic Library of Near Isogenic Lines. J. Am. Soc. Hortic. Sci. 132, 80–89 (2007).
Perin, C. et al. Genetic control of fruit shape acts prior to anthesis in melon (Cucumis melo L.). Mol. Genet. Genomics 266, 933–941 (2002).
Brown, P. J. et al. Distinct genetic architectures for male and female inflorescence traits of maize. PLOS Genet. 7, e1002383 (2011).
Wallace, J. G. et al. Association mapping across numerous traits reveals patterns of functional variation in maize. PLOS Genet. 10, e1004845 (2014).
Yano, K. et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927–936 (2016).
Cao, K. et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 7, 13246 (2016).
Tieman, D. et al. A chemical genetic roadmap to improved tomato flavor. Science (80-.). 355, 391–394 (2017).
Leplat, F., Pedas, P. R., Rasmussen, S. K. & Husted, S. Identification of manganese efficiency candidate genes in winter barley (Hordeum vulgare) using genome wide association mapping. BMC Genomics 17, 775 (2016).
Diaz, A. et al. Anchoring the consensus ICuGI genetic map to the melon (Cucumis melo L.) genome. Mol. Breed. 35, 1–7 (2015).
Clayberg, C. D. Interaction and linkage test of flesh colour genes in Cucumis melo L. Cucurbit Genet. Coop. 15, 53 (1992).
Astle, W. & Balding, D. Population structure and cryptic relatedness in genetic association studies. Stat. Sci. 24, 451–471 (2009).
Jefferey, C. A review of the Cucurbitaceae. Bot. J. Linn. Soc. 81, 233–247 (1980).
Larsson, S. J., Lipka, A. E. & Buckler, E. S. Lessons from Dwarf8 on the strengths and weaknesses of structured association mapping. PLoS Genet. 9, e1003246 (2013).
Huang, B. E. et al. MAGIC populations in crops: current status and future prospects. Theor. Appl. Genet. 128, 999–1017 (2015).
King, E. G., Macdonald, S. J. & Long, A. D. Properties and power of the Drosophila Synthetic Population Resource for the routine dissection of complex traits. Genetics 191, 935–949 (2012).
Dell’Acqua, M. et al. Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays. Genome Biol. 16, 167 (2015).
McMullen, M. D. et al. Genetic properties of the maize nested association mapping population. Science. 325, 737–741 (2009).
Pascual, L. et al. Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol. J. doi:10.1111/pbi.12282 (2014).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS One 9 (2014).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Endelman, J. B. & Jannink, J.-L. Shrinkage Estimation of the Realized Relationship Matrix. G3 Genes|Genomes|Genetics 2, 1405–1413 (2013).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, msw054 (2016).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B 57, 289–300 (1995).
Acknowledgements
Funding for this research was provided by the United States-Israel Binational Agricultural Research and Development Fund (BARD) US-4918-16CR, by the Israeli Ministry of Agriculture Chief Scientist grants no. 256-1103-15 and 277-0590-16 and in part by the ‘Center for the Improvement of Cucurbit Fruit Quality’, ARO, Israel. We appreciate and acknowledge Harry Paris for his critical language editing of the manuscript. Publication No. 201/2017 of the Agricultural Research Organization, Bet Dagan, Israel.
Author information
Authors and Affiliations
Contributions
A.G., J.B. and Y.T. conceived and designed the study. A.G., G.T., A.M., V.P. and U.S. performed field experiments and phenotyping. N.K., L.L. and A.A.S. provided genomic experimental support. A.G. analyzed the data. A.G. and Y.T. wrote the paper. All authors discussed the results, and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gur, A., Tzuri, G., Meir, A. et al. Genome-Wide Linkage-Disequilibrium Mapping to the Candidate Gene Level in Melon (Cucumis melo). Sci Rep 7, 9770 (2017). https://doi.org/10.1038/s41598-017-09987-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09987-4
This article is cited by
-
Charcoal rot (Macrophomina phaseolina) across melon diversity: evaluating the interaction between the pathogen, plant age and environmental conditions as a step towards breeding for resistance
European Journal of Plant Pathology (2022)
-
Identification of fruit firmness QTL ff2.1 by SLAF-BSA and QTL mapping in melon
Euphytica (2022)
-
Fine-mapping and identification of a candidate gene controlling seed coat color in melon (Cucumis melo L. var. chinensis Pangalo)
Theoretical and Applied Genetics (2022)
-
A cryptic variation in a member of the Ovate Family Proteins is underlying the melon fruit shape QTL fsqs8.1
Theoretical and Applied Genetics (2022)
-
Genetic characterization of melon accessions in the U.S. National Plant Germplasm System and construction of a melon core collection
Molecular Horticulture (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
