
Abstract
Populations of Streptococcus pneumoniae (SP) are typically structured into groups of closely related organisms or lineages, but it is not clear whether they are maintained by selection or neutral processes. Here, we attempt to address this question by applying a machine learning technique to SP whole genomes. Our results indicate that lineages evolved through immune selection on the groEL chaperone protein. The groEL protein is part of the groESL operon and enables a large range of proteins to fold correctly within the physical environment of the nasopharynx, thereby explaining why lineage structure is so stable within SP despite high levels of genetic transfer. SP is also antigenically diverse, exhibiting a variety of distinct capsular serotypes. Associations exist between lineage and capsular serotype but these can be easily perturbed, such as by vaccination. Overall, our analyses indicate that the evolution of SP can be conceptualized as the rearrangement of modular functional units occurring on several different timescales under different pressures: some patterns have locked in early (such as the epistatic interactions between groESL and a constellation of other genes) and preserve the differentiation of lineages, while others (such as the associations between capsular serotype and lineage) remain in continuous flux.
Similar content being viewed by others


Introduction
Streptococcus pneumoniae (the pneumococcus) is a gram-positive bacterial pathogen which, although commonly carried asymptomatically in the nasopharynx, can cause pneumonia, meningitis, septicemia and bacteremia in the young, elderly and immuno-compromised, being responsible for about 11% of worldwide deaths in children under 5 years of age1, 2. Populations of S. pneumoniae are antigenically diverse and can be stratified into more than 90 serotypes according to the antigenic properties of the expressed polysaccharide capsule, of which only 10–15 are responsible for most cases of invasive disease worldwide3. Reductions in disease rates have been achieved by the deployment of the PCV7 vaccine targeting 7 of the most common serotypes in invasive disease, and more recently through the use of PCV13 which extends coverage to an additional 6 serotypes. However this has been accompanied by an increase in the frequency of non-vaccine serotypes in many parts of the world, likely due to the removal of competition from vaccine serotypes4.
Like many other bacterial pathogen populations, S. pneumoniae may be organised into a number of so-called clonal complexes on the basis of allelic diversity at selected housekeeping loci (determining Multilocus Sequence Type5, 6,). Pneumococcal populations are also structured at a whole genome level into co-circulating lineages or Sequence Clusters (SC) bearing unique signatures of alleles7,8,9. The relationships between clonal complex, lineage and serotype are often found to be non-overlapping8, 10, although subject to perturbations such as through vaccination11.
The maintenance of discrete major lineages, and their associations with distinct serotypes and clonal complexes, is hard to ascribe to purely neutral processes, given the high rate of genetic exchange in these pathogen populations12, 13. We have previously proposed that extensive co-adaptation between loci may give rise to these patterns, as even small fitness differences among different combinations of alleles can lead to the loss of less fit genotypes under intense competition for resources14. Bacterial populations could also segregate into a set of successful metabolic types which are able to co-circulate by virtue of exploiting separate metabolic niches and thereby avoiding direct resource competition15. As an example, specific differences in the ability to absorb particular carbohydrate resources have been observed in functional genomics studies of S. pneumoniae 16, and these may reflect specialization upon different resources within the same environment as a means of avoiding competition. Establishing the contribution of co-adaptation and competition in the maintenance of discrete lineages is important since the outcome of certain interventions, such as vaccination, depends crucially on these underlying determinants of population structure17.
Here, we attempt to elucidate potential drivers of lineage structure by applying a machine learning technique known as the Random Forest Algorithm (RFA) to a dataset containing 616 whole genomes of S. pneumoniae collected in Massachusetts (USA) between 2001 and 20078. RFA-based methods have been robustly applied in genome-wide association studies of cancer and chronic disease risk18, species classification19, or in the search of viral determinants for host tropism, for instance by identifying the key amino-acid sites that determine host specificity of zoonotic viruses20, and by selecting the clear genetic distinctions in avian and human proteins of Influenza viruses21, 22. In the context of bacterial pathogens, these RFA-based and similar machine learning methods have been sucessfully used to analyse the genetic background of Escherichia coli cattle strains more likely to be virulent to humans23, to identify Staphylococcus aureus genetic variants associated with antibiotic resistance24, and to discover that repertoires of virulence proteins within different Legionella species are largely unique (non-overlapping)25. An RFA is an ensemble method that combines the information of multiple, regression or classification trees built around predictor variables towards a response variable. The output of an RFA is composed both of the classification success rates of the response variable and a ranking of the predictor variables quantifying their relative role in the classification process.
We used as response variables (i) the capsular serotype of each isolate (which had been determined by serological means), and (ii) the monophyletic Sequence Cluster (SC) to which samples had been assigned8. We set the predictor variables to be the 2135 genes for which we had obtained allelic profiles (effectively using a whole-genome multi-locus sequence typing approach, wgMLST)26 for each of the 616 isolates17. Using this method, we confirmed that capsular genes predict serotype, but found a clear disjunction between these genes and those which predict SC (lineage). Furthermore, our analyses revealed that, contrary to the expectations of neutrality, genes which predict lineage are non-randomly distributed across the genome, clustering within and around the groESL operon, leading us to propose that a combination of immune selection and coadaptation operating upon these loci may be the primary determinants of lineage structure.
Results
Classification success for serotype and sequence cluster
Classification of SC by the RFA was accurate (Fig. S1B) with all SC types being predicted with 100% success. This is a reflection of the strong correspondence between classification trees and taxonomy when based on genetic information, as explored in other studies19, and demonstrated by Austerlitz and colleagues when comparing the success of RFA, neighbour-joining and maximum-likelihood methodologies on simulated and empirical genetic data27.
By contrast, the success rate in identifying the capsular serotypes of the 616 whole genomes, although also very high (above 75% for the majority of serotypes), was not perfect (Fig. S1A). This is to be expected given the imperfect association between lineage and serotype, and also because certain serotypes were represented by very small numbers of isolates (as an extreme example, only a single isolate of serotype 21 was present and therefore classification success was nil).
The capsular locus is a strong predictor of serotype but performs indifferently in predicting sequence cluster
As might be expected, genes within the capsular locus (defined as being within but not including the genes dexB and aliA) were highly predictive of serotype, with their RFA scores appearing as outliers in the top 2.5% of the distribution defined by all 2135 genes in the dataset (Fig. 1A, see Methods). However, these did not score above average in predicting SC, as their RFA scores shifted closely to the distribution’s average (Fig. 1B). We noted, however, that many of these genes contained what appeared to be a high proportion of deletions across samples but, in fact, had not been matched with any known gene in the database (alleles ‘0’, see Methods) due to their high diversity at the level of the population (see, for example ref. 28). For certain genes, such as those encoding the polysaccharide polymerase Wzy and the flippase Wzx, the allelic notation process failed at least 50% of the time for over 90% of the isolates, essentially working only for serotype 23F (the reference genome) and the closely related 23A and 23B serotypes. In general, the degree of success in allelic notation of each gene was closely linked to the potential for alignment with its counterpart in the 23F reference genome (Fig. S4). Nonetheless, the same shift towards lower RFA scores of capsule-associated genes in predicting SC rather than serotype was observed upon performing a series of sensitivity classification exercises after excluding all genes which contained >50% (Fig. S2) or >10% (Fig. S3) of gene mismatches/deletions. When imposing an exclusion criterion of >10% we retained only the genes wze, wzg and wzh (in addition to two pseudogenes) within the capsular locus, and these could also clearly be seen to shift from above the upper 97.5% limit into the neutral expectation of RFA scores when predicting SC (Fig. S3).

Random forest classification. (A) Random forest analysis (RFA) for serotype classification. (A, top) Density function of RFA scores obtained for each gene in the dataset. The 95% boundaries are marked by the dashed lines. Small bars highlight the RFA scores of genes within particular groups (yellow for MLST genes, blue for capsular locus genes). (A, bottom) Genomic position for each gene in the dataset against their RFA score (normalised to [0,1]). The circular genome is presented in a linear form on the y-axis, with the first gene being dnaA and the last gene parB. MLST genes are marked in yellow diamonds (spi, xpt, glkA, aroE, ddlA, tkt) and genes within the capsular locus with blue diamonds (pseudogenes tagged with ‘x’). (B) RFA analysis for sequence cluster classification; figure details the same as in A. Blue shaded areas in both A and B subplots mark the capsular locus (genes within aliA and dexB).
We next performed the same analysis excluding all genes which showed mismatches or deletions above a threshold of 1%, in an attempt to eliminate possible biases in RFA output due residual information arising from the distribution of mismatches/deletions. This left us with 1581 genes which were shared by essentially all the samples in our dataset and for which function could be correctly ascertained by querying the reference genome. It is likely that these genes correspond to the approximately 1500 core cluster of orthologous genes (COGs) identified by Croucher et al. in their recent analysis of the same dataset7, although this could not be evaluated in detail given that this publication did not contain the list of COGs. This strict approach eliminated all of the genes considered above as belonging within the capsular locus, although flanking genes were retained and a number of these achieved the top 2.5% of RFA scores in predicting serotype (Fig. 2A, Table 1): 38% of the top genes occurred within 10 genes downstream and upstream of the capsular locus, and 90% were situated within 129 genes (which amounts for 6% of the genome). The remaining 10% of top-scoring genes, lytC, trpF, patB and SPN23F00400 were located at significantly longer distances from the capsular locus, at 963 (45% of the genome), 710, 469 and 270 (13% of the genome) genes away, respectively. None of the genes achieving the top 2.5% of RFA scores in predicting serotype (shown in red in Fig. 2) remained in the top 2.5% category when asked to predict SC. Similarly, all genes which achieved top scores in predicting SC (Table 2) were only of average importance in elucidating serotype (shown in green in Fig. 2).

Random forest classification excluding data with gene mismatches. (A) Random forest analysis (RFA) for serotype classification when excluding genes for which the allelic notation process had <99% positive matches with the reference genome. (A , top) Density function of RFA scores obtained for each gene in the dataset. The 95% boundaries are marked by the dashed lines. Small bars highlight the RFA scores of genes within particular groups (red for serotype, green for SC genes). (A, bottom) Genomic position for each gene in the dataset against their RFA score (normalised to [0,1]). The circular genome is presented in a linear form on the y-axis, with the first gene being dnaA and the last gene parB. Red and green diamonds mark the top 2.5% ranking genes for serotype and Sequence Cluster classification, respectively. (B) RFA for Sequence Cluster classification; figure details the same as in A. Blue shaded areas in both A and B mark the capsular locus (genes within aliA and dexB). Green shaded areas in both A and B mark the genes contiguous and including the groESL operon (Table 1).
The groESL operon is a strong predictor of sequence cluster
The majority of top-scoring genes for SC (75%) were randomly distributed along the genome (Fig. 2B), while 10 genes were found to be contiguous and contained within the groESL operon (clustering was statistically significant with p-value ≈ 1.52 × 10−06, Fig. S9). Notably, this operon, encoding the GroEL chaperone and GroES co-chaperone proteins (Table 2), has been reported in other studies to ascertain phylogeny and classification within the Streptococcus genus29 and between species of the S. viridans and S. mutans Streptococci groups30, 31.
A number of other top scoring genes in predicting SC have also previously been demonstrated to be powerful discriminators of genealogy in a range of bacterial species. For instance, sodA, encoding for the manganese superoxide dismutase, critical against oxidative stress and linked to both survival and virulence, has been highlighted in numerous studies for its relevance in identification of rare clones of pneumococci32, 33 and streptococci at the species level34, 35. Another example is the lmb gene, encoding for an extracellular protein with a key role in physiology and pathogenicity36, 37. Homologs of this protein have been documented to be present and discriminatory of at least 25 groups of the Streptococcus genus with possible similar functions38, 39.
The housekeeping genes included in multilocus sequence typing (MLST) classification performed no better than average in predicting SC across the sensitivity experiments (Figs 1 and S1–3). The exception was the Signal Peptidase I gene (spi), which featured in the top-scoring genes predicting SC under the strict 1% cutoff (Table 2). This is unsurprising, however, as MLST genes are unlikely to dictate lineage differentiation through selective processes, which endorses their choice as good discriminators of recent neutral diversification, in particular within recent epidemiological events5, 6.
Top-scoring genes for serotype are associated with resource competition and antibiotic resistance
When analyzing the 39 top-scoring, non-capsular genes which were highly predictive of serotype, we found 24 (62%) with compelling support for functional background that could mediate pneumococcal competitive interactions or niche specialization, at least in related streptococcal species (reviewed in detail in supplementary text). For instance, ATP-binding cassette (ABC) transporter genes, critical for intake, antibiotic resistance and metabolism, were found 5 times more frequently in the genes predictive of serotype compared to those determining SC (Tables 1 and 2). Notably, our approach selected the genes encoding for the pit ABC transporter, a key player in iron uptake known to exhibit strain-specific variation40, but did not select two other operons encoding iron transporters (piu, pia), which are conserved between S. pneumoniae strains40 and therefore unlikely to be predictors of serotype. Transport of essential substrates is also achieved by alternative systems which were also captured by our approach, such as the passive channel sodium symporter GlyP41 or the use of menaquinones and ubiquinones for electron transport (mevalonate pathway)42,43,44. We also found some of the top-scoring entries to be involved in functions associated with respiration (ecsA, mvaD, mvaK2) and amino acid, fatty acid and cell wall or capsular biosynthesis which amounted for approximately 25% of the top-scoring genes (trpF, fabG, lysC, mvaD, mvaK2, ritR, pbp1A, pbpX, mraW and mraY).
High RFA scores for serotype were also found among a number of genes flanking the capsular locus which are involved in antibiotic resistance, such as penicillin-binding protein genes pbpX and pbp1A, the 16S rRNA cytosine-methyltransferase gene mraW and the phospho-N-acetylmuramoyl-pentapeptide-transferase gene mraY. Genes involved in resistance to other antibiotics such as tomethicillin, vancomycin, daptomycin (vra operon)45 and the broad-spectrum quinolones family (patB)46,47,48 were also featured in the top-scoring genes. Also of note were entries linked to direct inter- and intra-species competition, either through factors related to immune escape or warfare. These included genes linked to pneumolysin expression and biofilm formation (luxS)49, 50, and production of bacteriocins (blpH)51, 52, ammonia (glmS)53 and lysozymes (lytC)54, 55.
Several top-scoring genes for SC classification are also key determinants of phenotype
The top-scoring genes predicting SC were discordant to the ones determining serotype and approximately 30% were found to have unknown functions (Table 2). However, we also found several examples of genes whose functions (reviewed in supplementary text) would be expected to be naturally linked with particular phenotypes such as virulence (sodA, lmb, pdhB, varZ, licA)32, 33, 36, 37, 56,57,58 or specific virulence traits such as host-cell adherence (pclA)59 or laminin binding (lmb)39. Several genes were also found to encode or directly produce proteins or protein-complexes which are highly immunogenic, such as the groEL 60,61,62, lmb 39, carB 63, 64, and licA 58, 65 genes.
Discussion
Our aim, in this paper, was to test the hypothesis that the stratification of pneumococcal populations into distinct sequence clusters or lineages occurred through neutral processes, with serotype diversity being superimposed upon the ensuing clonal framework to minimize antigenic interference between lineages. To this end, we applied a Random Forest Algorithm (RFA) to assess the contribution of different genes in determining the serotype or sequence cluster of isolates within a dataset containing 616 whole genomes of S. pneumoniae collected in Massachusetts (USA)8, for each of which we had obtained allelic profiles of 2135 genes of both known and unknown function17. By selecting the 2.5% of top RFA scores, we effectively focused on the subset of possibly selected units (genes) which present combinations of alleles that appear statistically more informative than expected at the genome level (see Methods for details). We show that by comparing the genomic localization and function of these top-scoring units (genes), general expectations concerning population structure can be revisited66, and inferences can be made concerning the evolutionary processes underlying the formation, relationship and maintenance of serotype and sequence cluster (lineage) at the population level.
Reassuringly, genes of the capsular locus (cps) and many of those flanking it achieved high RFA scores in predicting serotype. We also found a preponderance of genes scoring highly for serotype prediction to be associated with key functions that could define unique metabolic types that would have diversified in order to avoid direct resource competition, as previously proposed4, 17. However, 90% of the selected genes were at a distance of less than 6% of the genome to the cps locus and was therefore not possible to determine whether these had become segregated through competition or by physical (see e.g ref. 67) and/or functional associations with this locus. It should be noted that linkage disequilibrium is extremely high in this dataset (Fig. S7), and even if the selected genes had been found across the genome, it would be difficult to quantify the role of these genes in determining a metabolic type17.
Genes that were highly informative in predicting lineage (sequence cluster) were entirely distinct from those determining serotype. Contrary to what would be expected from a population structure maintained mostly by neutral processes, around a quarter of these genes co-localized within and around the groESL operon (marked with * in Table 2), which encodes the macromolecular machinery for a well-studied protein folding system centred around the chaperone GroEL and co-chaperone GroES68. In Escherichia coli, approximately 10% of total cytosolic proteins, including 67 essential proteins, have been demonstrated to have stable binding to GroEL, with 50 of these confirmed to depend on groESL folding via GroEL-depletion experiments69. GroEL is also known to be highly immunogenic in S. pneumoniae 61, 70, as well as in other bacterial species62, 71, 72. This raises the radically alternative possibility that sequence clustering may have arisen from immune selection operating on groEL in conjunction with extensive coadaptation with genes encoding the proteins which rely on this chaperonin system.
Classification success by our RFA approach was accurate for SC but was lower for serotype. Several factors may have influenced this discrepancy. For instance, the mean number of samples per serotype was lower than for SC (19.8 versus 38.5), providing in some cases very low levels of information per serotype to the machine learning technique. The capsular locus, where the majority of best predictors for serotype were located, also had the highest levels of allelic notation mismatches and crucial information may have been lost when applying the strict cutoff of 1%. In this context we note that the best predictors for serotype and lineage were found to be non-overlapping. This implies that capsular switches, assuming that only the cps had been switched, would not have affected the classification success of serotype (see ref. 8 for examples of switches in this dataset). On the other hand, switches involving genes that flank the cps could explain the lower success rates found for serotype. Dealing with the effects of these different factors, however, requires better sample representation per serotype, better annotation algorithms and detailed data on capsular switching events and in particular on the genes involved. At the moment such data are not available but achieving better serotype classification success is a possible line of future research.
Our results provide a mechanistic basis for the distinction proposed by Croucher and colleagues7, in the context of the same dataset, between infrequent macroevolutionary changes providing a stable backdrop for more frequent, and often transient, microevolutionary changes (see Fig. 3). The differentiation of the groESL operon would be a striking example of macroevolution, not only driving the emergence of S. pneumoniae sequence clusters but also serving to genealogically distinguish closely related bacterial species29,30,31. Several other genes scoring highly for SC were also found to encode or directly produce proteins or protein-complexes which are highly immunogenic (eg. lmb, carB, licA), and these may contribute to the maintenance of lineage structure by co-selection with groEL in accordance with the strain theory of host-pathogen systems in which immune selection operating on multiple immunogenic loci can cause the emergence of non-overlapping combinations of alleles75,76,77. In contrast, the emergence and maintenance of serotypes within major lineages would be dictated by differentiation in genes within and surrounding the capsular locus, and less permanent associations could arise between SC and serotype (at a microevolutionary scale) through resource competition14, 17 or indeed multi-locus immune selection operating on GroEL, the capsule, as well as other surface antigens74.

Population structure and vaccination. Conceptual representation of phylogenetic relationships between serotypes and Sequence Clusters (SC), where the former are defined by variation at the cps locus (arbitrarily designated X, W, Y, Z, M, and L, respectively coloured purple, yellow, green, orange, cyan and pink) and the latter are linked to variation in the groESL operon (arbitrarily designated A and B and respectively coloured red and blue). Circles symbolize genotypes, with size relative to their prevalence at the population level. Inner genome arcs represent epistatic links: those with the groESL operon extend across the genome, while links with the cps locus are more local. Within our framework and according to observed patterns8, serotypes will be dominantly associated with an SC. Current vaccine strategies (white inner area) that target a selection of capsular serotypes can lead to the expansion of non-vaccine serotypes (VISR73, 74), potentially within the same sequence cluster (VIMS17). Vaccine strategies based on groESL variants (grey area) would target entire lineages instead, including all uncommon serotypes within and thereby preventing their expansion.
We note that genes belonging to the Rec family are positioned in close proximity to both the contiguous clusters of top-scoring genes for SC and serotype (Tables 1 and 2). For example, the top-scoring gene recX is in close proximity to the groESL operon and encodes a regulatory protein that inhibits the RecA recombinase in multiple species of bacteria78,79,80,81. Restriction-modification systems (RMS) have been proposed as a means of maintaining species identity in a number of bacterial systems82 and this idea has been extended to the maintenance of lineages within meningococcal83 and pneumococcal7 species. Within our framework, RMS would act at even more local scale, principally to conserve the function of critical operons such as groESL, rather than prevent their recombination with other genes or operons. It has recently been demonstrated that GroEL in E. coli can be functionally replaced, at least partially, by an eukaryotic chaperonin84 indicating that the maintenance of particular associations of genes with the groESL operon is a consequence of their superior fitness rather than an inability to recombine. It is therefore tempting to speculate that RMS play a role in protecting the modularity of the genome and that population structure arises through selection favouring particular combinations of variants of these modules.
The proposed approach does not depend on the universal existence of the SCs described in the Croucher et al. dataset8. Instead, we rely on the fact that any whole genome dataset can be stratified and classified into major lineages by phylogenetic approaches. The research presented in the study is therefore intended to be a proof-of-principle under an ideal dataset for which SC and serotype classifications exist. Overall, our analyses support the hypothesis that lineage structure in maintained by co-adaptation and competition14 and show that these selection pressures converge upon the capsular locus and, surprisingly, the groESL operon. Our results endorse the development of vaccines against the associated chaperone protein, groEL, since targetting its protein folding machinery may provide a robust method (Fig. 3) of eliminating particular highly successful lineages rather than promoting the survival of those genotypes within it which carry cps loci encoding non-vaccine capsular serotypes17. We hope, for these reasons, that this work will stimulate further empirical testing of our hypothesis that immune selection against groEL may be a primary driver of lineage differentiation in the pneumococcus.
Methods
Sequence Data and Allelic Annotation
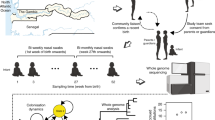
We used a dataset sequenced by Croucher et al., comprising 616 carriage S. pneumonaie genomes isolated in 2001, 2004 and 2007 from Massachusetts (USA). The data included 133, 203, 280 samples from 2001, 2004, 2007, respectively; and is stratified into 16 samples of serotype 10A, 50 of 11A, 7 of 14, 24 of 15A, 60 of 15BC, 8 of 16F, 5 of 17F, 6 of 18C, 73 of 19A, 33 of 19F, 1 of 21, 21 of 22F, 33 of 23A, 23 of 23B, 17 of 23F, 11 of 3, 4 of 31, 5 of 33F, 6 of 34, 49 of 35B, 18 of 35F, 2 of 37, 9 of 38, 47 of 6A, 17 of 6B, 33 of 6C, 3 of 7C, 11 of 7F, 4 of 9N, 6 of 9V and 14 of NT (see ref. 8 for collection details). Sequence reads were taken from the project ERP000889 on the European Nucleotide Archive (http://www.ebi.ac.uk/) and assembled using an automated pipeline with the Velvet algorithm17. In summary, we performed a whole genome multi-locus sequence typing (wgMLST) allelic notation26 using the BIGSdb software with an automated BLAST process85 and the Genome Comparator tool (with ATCC700669 serotype 23F, accession number FM211187, as the reference genome)17. This wgMLST approach resulted in the identification of 2135 genes in common between the reference and all the samples in the dataset. Alleles identical to the reference were classified as ‘1’, with subsequent sequences, differing at least by one base, labelled in increasing order. Genes were further classified as allele ‘0’ when genetic data present had no match to the genome of interest, or were found to be truncated or non-coding. For a visual representation of the allelic annotation and diversity please refer to S1 dataset of Watkins et al.17. Functional characterization of genes and gene families was done by literature search and access to the Kyotto Encyclopedia of Genes and Genomes (KEGG) database (www.genome.jpkeggpathway.html).The allelic matrix as obtained by this approach and used in the RFA analysis (see below) is herein made available in supplementary Table S1, which also includes the Accession Numbers, gene name, gene product, gene position in reference genome, and year of collection, Sequence Cluster and serotype of each sample.
Random Forest Approach
We implement a machine learning approach based on a Random Forest Algorithm (RFA) to predict particular features (serotype or Sequence Cluster) of each pneumococci isolate from information on the wgMLST allelic composition of the 2135 genes86. In summary, the RFA process takes the following pseudo-steps: (I) the response variable and predictor variables are chosen by the user; (II) a predefined number of independent bootstrap samples are drawn from the dataset with replacement, and a classification tree is fit to each sample containing roughly 2/3 of the data, for which predictor variable selection on each node split in the tree is conducted using only a small random subset of predictor variables; (III) the complete set of trees, one for each bootstrap sample, composes the random forest (RF), from which the status (classification) of the response variable is predicted as an average (majority vote) of the predictions of all trees. Compared to single classification trees, RFA increases prediction accuracy, since the ensemble of slight different classification results adjusts for the instability of the individual trees and avoids data overfitting87.
Here we use randomForest: Breiman and Cutler’s Random Forests for Classification and Regression, a software package for the R-statistical environment88. Predictor variables are set to be each gene in our genome samples and the response variable is set to the serotype or Sequence Cluster classification of each genome (as per ref. 8). We use the Mean Decrease Accuracy (MDA), or Breiman-Cutler importance, as a measure of predictor variable importance, for which classification accuracy after data permutation of a predictor variable is subtracted from the accuracy without permutation, and averaged over all trees in the RF to give an importance value87. The strategy herein employed is not of quantitative nature, as the absolute scale of scores produced by the RFA is dependent on the dataset being analyzed86. Instead, we focus on the 2.5% of top RFA scores as presented by the resulting MDA distribution for all genes, thus selecting the subset of genes which present combinations of alleles that appear statistically more informative than expected at the genome level (i.e. we assume that 95% of the scores should fall between the 2.5th and 97.5th percentiles). With this assumption and the approach detailed below, we effectively select the genes which present a p-value <0.05 given an intrinsic distribution of scores generated by data permutation (a null distribution of scores).
For the results presented in the main text, we assume the predictor variables to be numerical (as opposed to categorical). This assumption is known to introduce RF biases, as classification is effectively made by regression and artificial correlations between allele numbering and the features being selected (serotype and Sequence Cluster) may be present. The assumption is herein necessary since the RFA R-based implementation (version 3.6.12) has an upper limit of 53 categories per predictor variable and we find some genes to present up to 6 times this limit in allele diversity. The categorical constraint is a common feature of RFA implementations, as predictor variables with N categories imply 2N possible (binary) combinations for an internal node split, making the RFA method computationally impractical. Given this inherent RFA limitation, we implemented an input randomization strategy (random reassignment of values to alleles) to minimize potential bias. For this, M random permutations of each gene’s variant allelic numbering in the original dataset is performed, effectively creating M independent input matrices. The RFA is run over the input matrices and in the main results we present each gene’s average MDA score. Sensitivity analyses were performed by comparing RFA results between two independent sets of M = 50 input matrices (effectively comparing 100 independent runs) (Figs S5 and S10). Results suggest that the existing biases in independent runs of the RFA due to the assumption of numerical predictors are virtually mitigated with our input randomization strategy approach, specially for the experiments presented in the main results (i.e. using a 1% cutoff of gene mismatches, Fig. S10).
References
Song, J. Y. et al. Clinical and economic burden of invasive pneumococcal disease in adults: a multicenter hospital-based study. BMC infectious diseases 13, 202 (2013).
O’Brien, K. L. et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. The Lancet 374, 893–902 (2009).
Hausdorff, W. P., Feikin, D. R. & Klugman, K. P. Epidemiological differences among pneumococcal serotypes. Lancet Infectious Diseases 5, 83–93 (2005).
Cobey, S. & Lipsitch, M. Niche and neutral effects of acquired immunity permit coexistence of pneumococcal serotypes. Science (New York, N.Y.) 335, 1376–80 (2012).
Maiden, M. C. et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences of the United States of America 95, 3140–5 (1998).
Spratt, B. G. Multilocus sequence typing: Molecular typing of bacterial pathogens in an era of rapid DNA sequencing and the Internet. Current Opinion in Microbiology 2, 312–316 (1999).
Croucher, N. J. et al. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nature Communications 5, 1–12 (2014).
Croucher, N. J. et al. Population genomics of post-vaccine changes in pneumococcal epidemiology. Nature genetics 45, 656–63 (2013).
Cremers, A. J. H. et al. The post-vaccine microevolution of invasive Streptococcus pneumoniae. Scientific reports 5, 14952 (2015).
Brueggemann, A. B. et al. Clonal Relationships between Invasive and Carriage Streptococcus Pneumoniae and Serotype- and Clone-Specific Differences in Invasive Disease Potential. The Journal of Infectious Diseases 187, 1424–1432 (2003).
Beall, B. W. et al. Shifting genetic structure of invasive serotype 19A pneumococci in the United States. Journal of Infectious Diseases 203, 1360–1368 (2011).
Henriques-Normark, B., Blomberg, C., Dagerhamn, J., Bättig, P. & Normark, S. The rise and fall of bacterial clones: Streptococcus pneumoniae. Nature reviews. Microbiology 6, 827–37 (2008).
Fraser, C., Hanage, W. P. & Spratt, B. G. Neutral microepidemic evolution of bacterial pathogens. Proceedings of the National Academy of Sciences of the United States of America 102, 1968–73 (2005).
Buckee, C. O. et al. Role of selection in the emergence of lineages and the evolution of virulence in Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America 105, 15082–7 (2008).
Watkins, E. R., Maiden, M. C. & Gupta, S. Metabolic competition as a driver of bacterial population structure. Future Microbiology fmb–2016–0079 (2016).
Bidossi, A. et al. A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS ONE 7, e33320 (2012).
Watkins, E. R. et al. Vaccination Drives Changes in Metabolic and Virulence Profiles of Streptococcus pneumoniae. PLoS pathogens 11, e1005034 (2015).
Meng, Y. a., Yu, Y., Cupples, L. A., Farrer, L. A. & Lunetta, K. L. Performance of random forest when SNPs are in linkage disequilibrium. BMC Bioinformatics 10, 78 (2009).
Slabbinck, B., Waegeman, W., Dawyndt, P., De Vos, P. & De Baets, B. From learning taxonomies to phylogenetic learning: integration of 16S rRNA gene data into FAME-based bacterial classification. BMC bioinformatics 11, 69 (2010).
Aguas, R. & Ferguson, N. M. Feature Selection Methods for Identifying Genetic Determinants of Host Species in RNA Viruses. PLoS Computational Biology 9 (2013).
Eng, C. L. P., Tong, J. C. & Tan, T. W. Predicting host tropism of influenza A virus proteins using random forest. BMC Medical Genomics 7, S1–S1 (2014).
Eng, C. L. P., Tong, J. C. & Tan, T. W. Distinct host tropism protein signatures to identify possible zoonotic influenza a viruses. PLoS ONE 11, 1–12 (2016).
Lupolova, N., Dallman, T. J., Matthews, L., Bono, J. L. & Gally, D. L. Support vector machine applied to predict the zoonotic potential of E. coli O157 cattle isolates. Proceedings of the National Academy of Sciences 201606567 (2016).
Alam, M. T. et al. Dissecting vancomycin-intermediate resistance in staphylococcus aureus using genome-wide association. Genome Biology and Evolution 6, 1174–1185 (2014).
Burstein, D. et al. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nature Genetics 48, 167–175 (2016).
Klemm, E. & Dougan, G. Advances in Understanding Bacterial Pathogenesis Gained from Whole-Genome Sequencing and Phylogenetics. Cell Host and Microbe 19, 599–610 (2016).
Austerlitz, F. et al. DNA barcode analysis: a comparison of phylogenetic and statistical classification methods. BMC bioinformatics 10(Suppl 1), S10 (2009).
Bentley, S. D. et al. Putatively novel serotypes and the potential for reduced vaccine effectiveness: capsular locus diversity revealed among 5,405 pneumococcal genomes. Microbial Genomics 2 (2016).
Glazunova, O. O., Raoult, D. & Roux, V. Partial sequence comparison of the rpoB, sodA, groEL and gyrB genes within the genus Streptococcus. International Journal of Systematic and Evolutionary Microbiology 59, 2317–2322 (2009).
Teng, L.-j. et al. groESL Sequence Determination, Phylogenetic Analysis, and Species Differentiation for Viridans Group Streptococci groESL Sequence Determination, Phylogenetic Analysis, and Species Differentiation for Viridans Group Streptococci. Journal of Clinical Microbiology 40, 3172–3178 (2002).
Hung, W. C., Tsai, J. C., Hsueh, P. R., Chia, J. S. & Teng, L. J. Species identification of mutans streptococci by groESL gene sequence. Journal of Medical Microbiology 54, 857–862 (2005).
Obregón, V. et al. Molecular peculiarities of the lytA gene isolated from clinical pneumococcal strains that are bile insoluble. Journal of Clinical Microbiology 40, 2545–2554 (2002).
Arbique, J. C. et al. Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp. nov. Journal of Clinical Microbiology 42, 4686–4696 (2004).
Poyart, C., Quesne, G., Coulon, S., Berche, P. & Trieu-Cuot, P. Identification of streptococci to species level by sequencing the gene encoding the manganese-dependent superoxide dismutase. Journal of Clinical Microbiology 36, 41–47 (1998).
Martín-Galiano, A. J., Balsalobre, L., Fenoll, A. & De la Campa, A. G. Genetic characterization of optochin-susceptible viridans group streptococci. Antimicrobial Agents and Chemotherapy 47, 3187–3194 (2003).
Spellerberg, B. et al. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infection and Immunity 67, 871–878 (1999).
Terao, Y., Kawabata, S., Kunitomo, E., Nakagawa, I. & Hamada, S. Novel laminin-binding protein of Streptococcus pyogenes, Lbp, is involved in adhesion to epithelial cells. Infection and Immunity 70, 993–997 (2002).
Zhang, Y. M. et al. Prevalent distribution and conservation of streptococcus suis lmb protein and its protective capacity against the chinese highly virulent strain infection. Microbiological Research 169, 395–401 (2014).
Wahid, R. M. et al. Immune response to a laminin-binding protein (Lmb) in group a streptococcal infection. Pediatrics International 47, 196–202 (2005).
Jomaa, M. et al. Immunization with the iron uptake ABC transporter proteins PiaA and PiuA prevents respiratory infection with Streptococcus pneumoniae. Vaccine 24, 5133–5139 (2006).
Reizer, J., Reizer, A. & Saier, M. H. A functional superfamily of sodium/solute symporters. Biochimica et biophysica acta 1197, 133–66 (1994).
Wilding, E. I. et al. Identification, essentiality and evolution of the mevalonate pathway for isopentenyl diphosphate biosynthesis in {Gram}-positive cocci. J. Bacteriology 182, 4319–4327 (2000).
Buhaescu, I. & Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clinical Biochemistry 40, 575–584 (2007).
Holstein, S. A. & Hohl, R. J. Isoprenoids: Remarkable diversity of form and function. Lipids 39, 293–309 (2004).
Boyle-Vavra, S., Yin, S., Jo, D. S., Montgomery, C. P. & Daum, R. S. VraT/YvqF is required for methicillin resistance and activation of the VraSR regulon in Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 57, 83–95 (2013).
Garvey, M. I., Baylay, A. J., Wong, R. L. & Piddock, L. J. V. Overexpression of patA and patB, which encode ABC transporters, is associated with fluoroquinolone resistance in clinical isolates of Streptococcus pneumoniae. Antimicrobial Agents and Chemotherapy 55, 190–196 (2011).
El Garch, F. et al. Fluoroquinolones induce the expression of patA and patB, which encode ABC efflux pumps in Streptococcus pneumoniae. Journal of Antimicrobial Chemotherapy 65, 2076–2082 (2010).
Boncoeur, E. et al. PatA and PatB form a functional heterodimeric ABC multidrug efflux transporter responsible for the resistance of streptococcus pneumoniae to fluoroquinolones. Biochemistry 51, 7755–7765 (2012).
Joyce, E. A. et al. LuxS Is Required for Persistent Pneumococcal Carriage and Expression of Virulence and Biosynthesis Genes. Infection and Immunity 72, 2964–2975 (2004).
Xu, L. et al. Role of the luxS Quorum-Sensing System in Biofilm Formation and Virulence of Staphylococcus epidermidis Role of the luxS Quorum-Sensing System in Biofilm Formation and Virulence of Staphylococcus epidermidis. Infection and immunity 74, 488–496 (2006).
De Saizieu, A. et al. Microarray-based identification of a novel Streptococcus pneumoniae regulon controlled by an autoinduced peptide. Journal of Bacteriology 182, 4696–4703 (2000).
Reichmann, P. & Hakenbeck, R. Allelic variation in a peptide-inducible two-component system of Streptococcus pneumoniae. FEMS Microbiology Letters 190, 231–236 (2000).
Moye, Z. D., Burne, R. A. & Zeng, L. Uptake and metabolism of N-acetylglucosamine and glucosamine by Streptococcus mutans. Applied and Environmental Microbiology 80, 5053–5067 (2014).
García, P., González, M. P., García, E., García, J. L. & López, R. The molecular characterization of the first autolytic lysozyme of Streptococcus pneumoniae reveals evolutionary mobile domains. Molecular Microbiology 33, 128–138 (1999).
Eldholm, V., Johnsborg, O., Haugen, K., Ohnstad, H. S. & Havastein, L. S. Fratricide in Streptococcus pneumoniae: Contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology 155, 2223–2234 (2009).
Iacobone, M., Mantero, F., Basso, S. M., Lumachi, F. & Favia, G. Results and long-term follow-up after unilateral adrenalectomy for ACTH-independent hypercortisolism in a series of fifty patients. Journal of Endocrinological Investigation 28, 327–332 (2005).
Pancholi, V. & Chhatwal, G. S. Housekeeping enzymes as virulence factors for pathogens. International Journal of Medical Microbiology 293, 391–401 (2003).
Humphries, H. E. & High, N. J. The role of licA phase variation in the pathogenesis of invasive disease by Haemophilus influenzae type b. FEMS Immunology and Medical Microbiology 34, 221–230 (2002).
Paterson, G. K., Nieminen, L., Jefferies, J. M. C. & Mitchell, T. J. PclA, a pneumococcal collagen-like protein with selected strain distribution, contributes to adherence and invasion of host cells. FEMS Microbiology Letters 285, 170–176 (2008).
Kim, S. N., Kim, S. W., Pyo, S. N. & Rhee, D. K. Molecular cloning and characterization of groESL operon in Streptococcus pneumoniae. Mol Cells 11, 360–368 (2001).
Cao, J. et al. Protection against pneumococcal infection elicited by immunization with multiple pneumococcal heat shock proteins. Vaccine 31, 3564–3571 (2013).
Péchiné, S., Hennequin, C., Boursier, C., Hoys, S. & Collignon, A. Immunization using GroEL decreases Clostridium difficile intestinal colonization. PLoS ONE 8 (2013).
Svenson, S. B. & Lindberg, A. L. F. A. Artificial Salmonella Vaccines: Salmonella typhimurium 0- Antigen-Specific Oligosaccharide-Protein Conjugates Elicit Protective Antibodies in Rabbits and Mice. Immunity and infection 32, 490–496 (1981).
Kalynych, S., Morona, R. & Cygler, M. Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiology Reviews 38, 1048–1065 (2014).
Serino, L. & Virji, M. Phosphorylcholine decoration of lipopolysaccharide differentiates commensal Neisseriae from pathogenic strains: Identification of licA-type genes in commensal Neisseriae. Molecular Microbiology 35, 1550–1559 (2000).
McInerney, J. O. More than tree dimensions: Inter-lineage evolution’s ecological importance. Trends in Ecology and Evolution 28, 924–625 (2013).
Enright, M. C. & Spratt, B. G. Extensive variation in the ddl gene of penicillin-resistant Streptococcus pneumoniae results from a hitchhiking effect driven by the penicillin-binding protein 2b gene. Molecular biology and evolution 16, 1687–95 (1999).
Hayer-Hartl, M., Bracher, A. & Hartl, F. U. The GroEL-GroES Chaperonin Machine: A Nano-Cage for Protein Folding (2016).
Kerner, M. J. et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 122, 209–220 (2005).
Khan, M. N., Shukla, D., Bansal, A., Mustoori, S. & Ilavazhagan, G. Immunogenicity and protective efficacy of GroEL (hsp60) of Streptococcus pneumoniae against lethal infection in mice. FEMS Immunology and Medical Microbiology 56, 56–62 (2009).
Hennequin, C. et al. GroEL (Hsp60) of Clostridium difficile is involved in cell adherence. Microbiology 147, 87–96 (2001).
Wuppermann, F. N., Mölleken, K., Julien, M., Jantos, C. A. & Hegemann, J. H. Chlamydia pneumoniae GroEL1 protein is cell surface associated and required for infection of HEp-2 cells. Journal of Bacteriology 190, 3757–3767 (2008).
Chang, Q. et al. Stability of the pneumococcal population structure in Massachusetts as PCV13 was introduced. BMC infectious diseases 15, 68 (2015).
Croucher, N. J. et al. Selective and Genetic Constraints on Pneumococcal Serotype Switching. PLoS Genetics 11, 1–21 (2015).
Gupta, S. Chaos, Persistence, and Evolution of Strain Structure in Antigenically Diverse Infectious Agents. Science 280, 912–915 (1998).
Gupta, S. et al. The maintenance of strain structure in populations of recombining infectious agents. Nature medicine 2, 437–42 (1996).
Lourenço, J., Wikramaratna, P. S. & Gupta, S. MANTIS: an R package that simulates multilocus models of pathogen evolution. BMC bioinformatics 16, 176 (2015).
Bergé, M., Mortier-Barrière, I., Martin, B. & Claverys, J. P. Transformation of Streptococcus pneumoniae relies on DprA- and RecA-dependent protection of incoming DNA single strands. Molecular Microbiology 50, 527–536 (2003).
Venkatesh, R. et al. RecX protein abrogates ATP hydrolysis and strand exchange promoted by RecA: insights into negative regulation of homologous recombination. Proceedings of the National Academy of Sciences of the United States of America 99, 12091–12096 (2002).
Stohl, E. A. et al. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. Journal of Biological Chemistry 278, 2278–2285 (2003).
Galvão, C. W. et al. The RecX protein interacts with the RecA protein and modulates its activity in herbaspirillum seropedicae. Brazilian Journal of Medical and Biological Research 45, 1127–1134 (2012).
Jeltsch, A. Maintenance of species identity and controlling speciation of bacteria: A new function for restriction/modification systems? Gene 317, 13–16 (2003).
Budroni, S. et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proceedings of the National Academy of Sciences of the United States of America 108, 4494–9 (2011).
Shah, R. et al. Replacement of GroEL in Escherichia coli by the Group II Chaperonin from the Archaeon Methanococcus maripaludis. Journal of Bacteriology 198, 2692–2700 (2016).
Altschul, S., Gish, W., Miller, W., Myers, E. & Lipman, D. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990).
Breiman, L. Random forests. Machine Learning 45, 5–32 (2001).
Friedman, J., Hastie, T. J. & Tibshirani, R. The Elements of Statistical Learning. Springer Series in Statistics (Springer New York, New York, NY, 2001).
Liaw, a & Wiener, M. Classification and Regression by randomForest. R news 2, 18–22 (2002).
Acknowledgements
The authors acknowledge the sequence data kindly given by Angela Brueggemann and Andries van Tonder, and Richard Moxon for the valuable comments on a previous version of this manuscript. This research was funded by the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. 268904 - DIVERSITY (www.erc.europa.eu; www.royalsociety.org). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.L. and S.G. designed the study. J.L. performed the experiments and wrote the initial manuscript. J.L., E.W.R., S.G. and M.C.J.M. revised the manuscript. J.L., U.O., S.J.P. and C.M. curated the data and revised gene functionality.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lourenço, J., Watkins, E.R., Obolski, U. et al. Lineage structure of Streptococcus pneumoniae may be driven by immune selection on the groEL heat-shock protein. Sci Rep 7, 9023 (2017). https://doi.org/10.1038/s41598-017-08990-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08990-z
This article is cited by
-
Reverse immunodynamics: a new method for identifying targets of protective immunity
Scientific Reports (2019)
-
Identifying genes associated with invasive disease in S. pneumoniae by applying a machine learning approach to whole genome sequence typing data
Scientific Reports (2019)
-
Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
