
Abstract
Autosomal dominant non-syndromic hearing loss (ADNSHL) is genetically heterogeneous with more than 35 genes identified to date. Using a massively parallel sequencing panel targeting 159 deafness genes, we identified a novel missense variant of POU4F3 (c.982A>G, p.Lys328Glu) which co-segregated with the deafness phenotype in a three-generation Taiwanese family with ADNSHL. This variant could be classified as a “pathogenic variant” according to the American College of Medical Genetics and Genomics guidelines. We then performed subcellular localization experiments and confirmed that p.Lys328Glu compromised transportation of POU4F3 from the cytoplasm to the nucleus. POU3F4 p.Lys328Glu was located within a bipartite nuclear localization signal (NLS), and was the first missense variant in bipartite NLS of POU4F3 validated in functional studies. These findings expanded the mutation spectrum of POU4F3 and provided insight into the pathogenesis associated with aberrant POU4F3 localization.
Similar content being viewed by others


Introduction
Autosomal dominant non-syndromic hearing loss (ADNSHL) is a heterogeneous disease entity with more than 35 genes identified to date (http://hereditaryhearingloss.org/). From an epidemiological perspective, none of these ADNSHL genes is more prevalent than another, making it difficult to perform genetic testing using conventional Sanger sequencing1. Recently, massively parallel sequencing (MPS), also known as targeted next-generation sequencing (NGS), has been proven to be a powerful tool in addressing genetically heterogeneous hereditary hearing impairment2. By using MPS-based panels, genetic causes could be determined in >50% of cases with ADNSHL3.
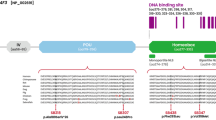
POU4F3 (MIM #602460) is one of the earliest deafness genes identified to cause ADNSHL DFNA15. In 1998, Vahava et al. detected an 8-base pair deletion in POU4F3 in a five-generation Israeli Jewish family4. To date, only 12 POU4F3 causative variants have been reported in the literature, that is, six missense variants5,6,7,8,9,10, five frameshift deletions4, 11,12,13,14, and a large deletion encompassing the entire gene15, 16. POU4F3 is located on 5q31 and encodes a protein of 338 amino acids, which functions as a transcription factor with two DNA-binding domains: the POU-specific domain and the POU homeodomain17. Two nuclear localization signals (NLSs) crucial for active protein transport into the nucleus are located within the POU homeodomain. The first is a monopartite NLS located at amino acids 274 to 278, and the second is a bipartite NLS located at amino acids 314 to 33118. According to previous reports, mutations in POU4F3 might lead to late-onset bilateral progressive hearing loss with down-sloping audiometric configurations8.
Results
Clinical features
The proband of the family was a 45-year-old woman who had bilateral progressive hearing impairment since around 30 years of age. Audiogram of the better ear of the proband showed the following hearing acuity: 40 dB at 250 Hz, 80 dB at 500 Hz, 90 dB at 1000 Hz, 115 dB at 2000 Hz, 120 dB at 4000 Hz, and 110 dB at 8000 Hz. Audiometry showed a down-sloping configuration (Fig. 1b). Her grandfather, her mother, and her mother’s two half-sisters (with the same father) also presented with late onset progressive hearing impairment (Fig. 1a).

(a) The pedigree and segregation pattern of the family, which harbored POU4F3 p.Lys328Glu. (b) The audiogram of both ears in the proband of the family revealed profound hearing loss with down-sloping shape. Hearing levels of the right and the left ear are marked using red and blue lines, respectively. (c) MPS-based panel identified a POU4F3 c.982A>G, p.Lys328Glu variant in the proband and this variant was validated by Sanger sequencing.
Identification of the causative variant
Using MPS-based panel, the average depth of coverage reached 134 folds, with 99.7% of sequences having coverage greater than one fold, 90.2% greater than 30 folds, and 63.8% greater than 100 folds. There were 333 variants located within exons or splice sites of the targeted 159 genes, nearly half of them were synonymous (166 out of 333). After excluding synonymous variants, there were nine variants, including seven nonsynonymous variants and two non-frameshift deletions, with allele frequencies less than 0.5% in the NHLBI-ESP 6500 exome project, 1000 Genomes project, and the East Asian population of the ExAC project (Supplementary Table S1). Prediction of pathogenicity using seven algorithms concluded that POU4F3 p.Lys328Glu (c.982A>G) was the only candidate variant that matched the inheritance pattern.
The c.982A>G is in exon 2 of POU4F3 (NM_002700.2). We performed Sanger sequencing (Fig. 1c) and found POU4F3 c.982A>G co-segregated with the deafness phenotype in the family (Fig. 1a). POU4F3 c.982A>G was absent in the NHLBI-ESP 6500 exome project, 1000 Genomes project, ExAC project, 100 normal-hearing Han Chinese controls, and Taiwan Biobank database. All seven of the algorithms reported deleterious effects of the POU4F3 p.Lys328Glu.
Effect of p.Lys328Glu on subcellular localization of POU4F3
The p.Lys328Glu is located within the bipartite NLS of the conserved POU homeodomain (Fig. 2a), suggesting the variant might affect the subcellular localization of POU4F318. To confirm this, we expressed the wild-type or mutant POU4F3 in COS-7 cells. As shown in Fig. 3, whereas the wild-type POU4F3 was located almost exclusively in the nucleus, a portion of the mutant POU4F3 was retained in the cytoplasm, indicating the localization ability of the mutant protein was compromised. Quantification of the transfected cells revealed that the percentage of cells with mutant POU4F3 in the cytoplasm (88%) was much higher than that of cells with the wild-type protein in the cytoplasm (19%).

(a) The POU4F3 p.Lys328 indicated in red shading was located within the bipartite nuclear localization signal (NLS) in the POU homeodomain. The bipartite NLS was conserved across nine species. (b) The location of six reported POU4F3 missense variants and p.Lsy328Glu. POU-specific domain and POU homeodomain are indicated by black boxes. Two NLSs were indicated by red lines. (c) The predicted protein products of five reported POU4F3 frameshift variants. Boxes with slash indicate the new amino acid sequences after the frameshift.

The POU4F3 c.982A>G mutation altered the subcellular localization of transcription factor POU4F3. Nuclei stained with DAPI (blue) and POU4F3 detected by Anti-DYKDDDDK-Tag antibody (red) were visualized by confocal microscopy. The white arrows indicate POU4F3 outside the nuclei.
Pathogenicity of POU4F3 p.Lys328Glu
When classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines19, POU4F3 p.Lys328Glu was predicted as deleterious by multiple in silico algorithms, meeting the PP3 category; it co-segregated with deafness in the family, fulfilling the PP1 category; it was absent from controls in multiple population databases and located in a critical functional domain, thus fitting the PM2 and PM1 categories, respectively; and most importantly, our in vitro functional studies demonstrated that this variant compromised protein function, fulfilling the PS3 category. In summary, POU4F3 p.Lys328Glu met five criteria, namely, PS3, PM1, PM2, PP1, and PP3, and could be classified as a “pathogenic variant” according to the ACMG guidelines.
Prevalence of POU4F3 variants in hearing-impaired families
To determine whether POU4F3 variants cause deafness in other Taiwanese families, we sequenced both exons of POU4F3 in 13 additional unrelated families with ADNSHL by either Sanger sequencing or the MPS panel. None of these families harbored causative variants in POU4F3. Similarly, we did not identify any causative variants of POU4F3 in additional 210 hearing-impaired families, which have been subjected to our MPS-based panel for genetic testing, that is, 30 autosomal recessive, four X-linked, and 176 sporadic families. In another paper describing the MPS results of 1119 patients with hearing loss, POU4F3 was not included in the 49 genes in which pathogenic variants were detected3. In other words, variants in POU4F3 are a rare cause of deafness from an epidemiological perspective.
Discussion
In this study, we identified a novel missense variant POU4F3 p.Lys328Glu causing ADNSHL in a Taiwanese family. Functional experiments was performed to evaluate the pathogenesis of the mutant protein, and the mislocalization of the protein was detected.
Among the 13 POU4F3 causative variants documented in the literature, POU4F3 p.Lys328Glu was the second missense mutation identified within the bipartite NLS (amino acids 314–331) of the POU homeodomain (Fig. 2b,c). Prior to this study, Kim et al. reported p.Arg326Lys in a Korean family with late-onset ADNSHL, but no functional studies were performed to examine the effect of p.Arg326Lys on the subcellular localization of POU4F38. Previous in vitro experiments have demonstrated defected nuclear localization of POU4F3 if variants result in a truncated protein lacking the bipartite NLS alone18 or both mono- and bipartite NLSs11. Our findings further add that missense variants involving key amino acid residues within NLSs might also affect POU4F3 protein trafficking.
Monopartite NLSs contain one cluster of basic amino acids, whereas bipartite NLSs are composed of two clusters of basic amino acids separated by a linker20. POU4F3 contains both mono- and bipartite NLSs: the sequence of the monopartite NLS is RKRKR, and the sequence of the bipartite NLS is KKNVVRVWFCNLQRQKQKR18. Weiss et al. demonstrated that the first two (KK) and last two (KR) amino acids of bipartite NLS were crucial for the nuclear localization of POU4F3. The causative missense variant p.Lys328Glu identified in this study corresponded to the fourth last amino acid of the bipartite NLS. It is conceivable that the replacement of a basic lysine with an acidic glutamate significantly changes the molecular characteristics of the bipartite NLS, indicating that correct alignment of the basic amino acids clusters within NLSs is essential for protein localization.
In addition to mislocalization, mutations in POU4F3 might also affect protein stability. Weiss et al. revealed that POU4F3 is a very short-lived protein and the half-life of the mutant protein (p.Ile295Thrfs*5) is longer than that of the wild-type protein18. Another study by Collin et al. examined the stability of two mutant POU4F3 proteins with missense variants, p.L223P and p.L289F. The stability of these mutant proteins was not different from that of the wild-type protein5. Recent studies have demonstrated a novel function of NLS in regulating protein stability through ubiquitin/proteasome system21, 22. Deletion of NLS containing lysine ubiquitination sites could decrease protein degradation21. The p.Ile295Thrfs*5 mutation disrupted the bipartite NLS of POU4F3, whereas p.L223P and p.L289F did not. This may explain the extended half-life of POU4F3 with the p.Ile295Thrfs*5 mutation. The p.Lys328Glu mutation identified in this study may also affect POU4F3 degradation, because of the substitution of a lysine residue for a glutamic acid residue within the NLS. Further studies are needed to elucidate the effects of different mutations on the stability of POU4F3.
The pathogenetic mechanisms underlying hearing impairment of patients with POU3F4 variants remain unclear. The molecular mechanisms of dominant inheritance includes haploinsufficiency, gain of function and dominant-negative effect23. As a dominant-negative effect has been ruled out, haploinsufficiency is the most likely mechanism so far5, 18. Although heterozygous Pou4f3 knockout mice exhibited normal hearing comparable to wild-type mice24, the mechanism of haploinsufficiency has been supported by two different studies. Deletion of the entire POU4F3 has been reported in a Brazilian family with ADNSHL15. A Japanese study also identified a POU4F3 frameshift variant (c.1007del), which would produce a transcript without in-frame stop codon (p. Ala336Valfs*? in Fig. 2c)12, and presumably the nonstop mRNAs might be degraded through non-stop decay25. In other words, both variants caused the loss of one copy of POU4F3, indicating the mechanism of haploinsufficiency. The subcellular protein mislocalization shown in this study and others5, 11, 18 also supports the mechanism of haploinsufficiency.
In conclusion, by using an MPS-based genetic testing panel targeting 159 known deafness genes, we identified a novel POU4F3 pathogenic variant, p.Lys328Glu, in a Taiwanese family with ADNSHL. POU4F3 p.Lys328Glu interrupted the bipartite NLS and prevented the transportation of POU4F3 from the cytoplasm to the nucleus. These findings expanded the mutation spectrum of the rare deafness gene POU4F3, and provided insights into the pathogenetic mechanisms associated with aberrant POU4F3 localization.
Methods
Subjects and clinical evaluation
A three-generation deafness family with five affected members was recruited in the study (Fig. 1a). Comprehensive family history; previous medical records; and results of physical, neurological, audiological examinations were obtained and analyzed. Audiological results were characterized with respect to two parameters, namely, hearing levels and audiogram shapes26. Hearing level of the better ear, which was calculated using a 4-tone average (0.5, 1, 2, and 4 kHz), was labeled as mild (20–40 dBHL), moderate (41–70 dBHL), severe (71–95 dBHL), or profound ( > 95 dBHL) hearing loss (GENDEAF: http://audiology.unife.it/www.gendeaf.org/index.html). Informed consent was obtained from all participants and all the procedures used in the study were approved by the Research Ethics Committee of the National Taiwan University Hospital. All methods were performed in accordance with the relevant guidelines and regulations.
Targeted MPS-based deafness panel
Genomic DNA of the proband was extracted from peripheral blood and subjected to an MPS-based deafness panel targeting 159 known deafness genes. The sequencing methods and analysis pipelines have been previously reported27, 28. In brief, genomic DNA was fragmented into 800 bps. After sample preparation, DNA fragments were enriched using custom probes designed to capture 1,299,144 base pairs with target regions encompassing 3,647 coding and non-coding exons of 159 deafness genes. Paired-end sequencing was performed by the Illumina Miseq platform (Illumina Inc., San Diego, CA, USA), which produced 300 bps reads.
Data analysis and filtering
We used BWA-MEM, Picard, GATK, and ANNOVAR to perform reads mapping, converting, sorting, variants calling, and annotation. Data filtering was conducted by an in-house perl script with the following steps: selection of variants located in the targeted 159 genes; filtering out of variants with allele frequencies more than 0.5% in the NHLBI-ESP 6500 exome project (http://evs.gs.washington.edu/EVS/), 1000 Genomes project (http://www.1000genomes.org/), Exome Aggregation Consortium (ExAC) projects (http://exac.broadinstitute.org/), 100 normal-hearing Han Chinese subjects, and Taiwan Biobank database (https://taiwanview.twbiobank.org.tw/); prediction of the pathogenicity of retained missense variants by seven algorithms including PolyPhen-2, SIFT, LRT, MutationTaster, MutationAssessor, FATHMM, and MetaLR; and confirmation that candidate variants are located in conserved regions of nine species and co-segregate with the deafness phenotype.
Sanger sequencing
Genomic DNA was extracted from peripheral blood or saliva samples of four affected members and two unaffected members. Sanger sequencing was performed to validate the variants identified by the MPS panel and to examine the co-segregation of variants with deafness among the family members. Allele frequencies of variants segregating with the phenotype were also verified in a panel of 100 normal-hearing Han Chinese subjects.
Cell transfection and immunocytochemistry
COS-7 cells were transiently transfected by a lentivirus with constructs encoding either the wild-type or the mutant fusion protein pWPT-POU4F3-6XHis-Flag. Transfected COS-7 cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and blocked in 10% goat serum. After incubation with DYKDDDDK Tag Antibody (Alexa Fluor 594 Conjugate, Thermo Fisher Scientific, Waltham, MA, USA), the samples were examined with a laser scanning confocal microscope (Zeiss LSM 510, Carl Zeiss, Germany). For quantitative analysis, 80-100 cells with mutant or wild-type POU4F3 mislocalization were counted, and the percentage of cells expressing POU4F3 in the cytoplasm was calculated.
References
Hilgert, N., Smith, R. J. & Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat. Res. 681, 189–196 (2009).
Shearer, A. E. & Smith, R. J. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss: The New Standard of Care. Otolaryngol. Head Neck Surg. 153, 175–182 (2015).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450 (2016).
Vahava, O. et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279, 1950–1954 (1998).
Collin, R. W. et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 29, 545–554 (2008).
Pauw, R. J. et al. Audiometric characteristics of a Dutch family linked to DFNA15 with a novel mutation (p.L289F) in POU4F3. Arch. Otolaryngol. Head Neck Surg. 134, 294–300 (2008).
Baek, J. I. et al. Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J. Rare Dis. 7, 60 (2012).
Kim, H. J. et al. SNP linkage analysis and whole exome sequencing identify a novel POU4F3 mutation in autosomal dominant late-onset nonsyndromic hearing loss (DFNA15). PLoS One 8, e79063 (2013).
Miyagawa, M., Naito, T., Nishio, S. Y., Kamatani, N. & Usami, S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS One 8, e71381 (2013).
Wei, Q. et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J. Transl. Med. 12, 311 (2014).
Lee, H. K., Park, H. J., Lee, K. Y., Park, R. & Kim, U. K. A novel frameshift mutation of POU4F3 gene associated with autosomal dominant non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 396, 626–630 (2010).
Mutai, H. et al. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: a cross-sectional, multi-center next-generation sequencing study. Orphanet J. Rare Dis. 8, 172 (2013).
Yang, T., Wei, X., Chai, Y., Li, L. & Wu, H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J. Rare Dis. 8, 85 (2013).
Cai, X. Z. et al. Exome sequencing identifies POU4F3 as the causative gene for a large Chinese family with non-syndromic hearing loss. J. Hum. Genet. 62, 317–320 (2017).
Freitas, E. L. et al. Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. Eur. J. Med. Genet. 57, 125–128 (2014).
Rosenberg, C. et al. Clinical Genetics Genomic copy number alterations in non-syndromic hearing loss. Clin. Genet. 89, 473–477 (2015).
Gerrero, M. R. et al. Brn-3.0: a POU-domain protein expressed in the sensory, immune, and endocrine systems that functions on elements distinct from known octamer motifs. Proc. Natl. Acad. Sci. USA. 90, 10841–10845 (1993).
Weiss, S. et al. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol. Cell. Biol. 23, 7957–7964 (2003).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Robbins, J., Dilworth, S. M., Laskey, R. A. & Dingwall, C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell 64, 615–623 (1991).
Hutchins, E. J., Belrose, J. L. & Szaro, B. G. A novel role for the nuclear localization signal in regulating hnRNP K protein stability in vivo. Biochem. Biophys. Res. Commun. 478, 772–776 (2016).
An, L. et al. Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl. Acad. Sci. USA 114, E2872–E2881 (2017).
Wilkie, A. O. The molecular basis of genetic dominance. J. Med. Genet. 31, 89–98 (1994).
Keithley, E. M., Erkman, L., Bennett, T., Lou, L. & Ryan, A. F. Effects of a hair cell transcription factor, Brn-3.1, gene deletion on homozygous and heterozygous mouse cochleas in adulthood and aging. Hear. Res. 134, 71–76 (1999).
Klauer, A. A. & van Hoof, A. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip Rev RNA 3, 649–660 (2012).
Wu, C. C., Chen, Y. S., Chen, P. J. & Hsu, C. J. Common clinical features of children with enlarged vestibular aqueduct and mondini dysplasia. Laryngoscope 115, 132–137 (2005).
Lin, Y. H. et al. Identification of a novel GATA3 mutation in a deaf Taiwanese family by massively parallel sequencing. Mutat. Res. 771, 1–5 (2015).
Wu, C. C. et al. Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine 94, e1073 (2015).
Acknowledgements
This study was supported by research grants from the Ministry of Science and Technology of the Executive Yuan of Taiwan (MOST 100-2314-B-002-031-MY3). We would like to thank National Research Program for Biopharmaceuticals (NRPB, MOST 104-2325-B-492-001) and National Center for High-performance Computing (NCHC) of National Applied Research Laboratories (NARLabs) of Taiwan for providing computational resources and storage resources. We thank the NTUH A1 Center of Genetic Testing for their experiments and technical support. We also wish to thank all subjects and their parents for participating in the present study.
Author information
Authors and Affiliations
Contributions
C.J.H., P.L.C., and C.C.W. conceived and designed the experiments. Yin-Hung, L., Yi-Hsin, L., and Y.C.L. performed the experiments. T.C.L., C.J.H., and C.C.W. collected the data. Yin-Hung, L., and C.Y.C. analyzed the data. Yin-Hung, L., P.L.C., and C.C.W. drafted manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, YH., Lin, YH., Lu, YC. et al. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci Rep 7, 7551 (2017). https://doi.org/10.1038/s41598-017-08236-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08236-y
This article is cited by
-
Highly variable hearing loss due to POU4F3 (c.37del) is revealed by longitudinal, frequency specific analyses
European Journal of Human Genetics (2023)
-
An integrative approach for pediatric auditory neuropathy spectrum disorders: revisiting etiologies and exploring the prognostic utility of auditory steady-state response
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
