
Abstract
MAS solid-state NMR is capable of determining structures of protonated solid proteins using proton-detected experiments. These experiments are performed at MAS rotation frequency of around 110 kHz, employing 0.5 mg of material. Here, we compare 1H, 13C correlation spectra obtained from protonated and deuterated microcrystalline proteins at MAS rotation frequency of 111 kHz, and show that the spectral quality obtained from deuterated samples is superior to those acquired using protonated samples in terms of resolution and sensitivity. In comparison to protonated samples, spectra obtained from deuterated samples yield a gain in resolution on the order of 3 and 2 in the proton and carbon dimensions, respectively. Additionally, the spectrum from the deuterated sample yields approximately 2–3 times more sensitivity compared to the spectrum of a protonated sample. This gain could be further increased by a factor of 2 by making use of stereospecific precursors for biosynthesis. Although the overall resolution and sensitivity of 1H, 13C correlation spectra obtained using protonated solid samples with rotation frequencies on the order of 110 kHz is high, the spectral quality is still poor when compared to the deuterated samples. We believe that experiments involving large protein complexes in which sensitivity is limiting will benefit from the application of deuteration schemes.
Similar content being viewed by others


Introduction
Magic Angle Spinning (MAS) solid-state NMR for biological samples has taken a large leap forward during the past years1,2,3,4,5,6,7,8. Advancements in NMR technology such as the development of ultrahigh magnetic fields (ω0/2π > 1 GHz), emergence of ultrafast spinning probes (ωr/2π > 100 kHz), isotopic labelling strategies and development of new NMR methods are some of the key points behind this success. Solid-state NMR samples are spun in rotors at the magic angle (Arctan [√2] = 54.74°) with respect to the main magnetic field. The maximum achievable frequency of sample spinning at the magic angle is inversely proportional to the diameter of the solid-state NMR rotor9. Therefore, faster spinning rotors imply smaller volumes with less amount of sample and thus, a reduced sensitivity of the NMR spectra to start out with. Other parameters such as the diameter of the rf coil, detection of protons instead of heteronuclei may counteract the decrease of sample volume10.
Protons yield the highest sensitivity for detection due to their high gyromagnetic ratio. However, the strong proton-proton dipole couplings yield significant line broadening. Using deuterated samples helps to recover the spectral resolution while retaining high sensitivity. Several deuteration strategies have been proposed for solids during the last decade, that are now widely used. In general, deuteration strategies have to be adopted according to the available NMR hardware, in particular the achievable MAS frequency. Specific labelling schemes allow to study exchangeable as well as non-exchangeable protons. At a maximum MAS frequency of 24 kHz (3.2 mm MAS rotor, 10–20 mg sample), partial deuteration (H:D ≈ 20:80) at exchangeable amide sites in an extensively deuterated environment is required to obtain optimal spectral quality11,12,13. The emergence of 1.3 mm probes (2 mg of sample, maximum ~60 kHz MAS) allowed to employ 100% protons at exchangeable sites in an otherwise deuterated sample without having to sacrifice sensitivity or resolution14,15,16,17. On the other hand, the proton density in the aliphatic sidechain is significantly higher compared to the amide backbone. For such non-exchangeable protons, several fractional deuteration protocols have been implemented for solid-state NMR such as random deuteration at the aliphatic sites or RAP18, selective ILV-methyl labelling19, SAIL20, iFD21 and proton-cloud22 schemes. It was shown that fractional labelling of side chain protons yields excellent spectral resolution even at moderate MAS frequencies23,24,25,26,27. Finding the optimal deuteration scheme for non-exchangeable sites at a given MAS frequency poses a greater challenge in comparison to backbone amides, since deuteration itself limits the observation of side chain resonances. In addition, deuteration of a protein often significantly reduces protein expression yields28.
It has been suggested recently that sample rotation with a frequency of ~110 kHz (in 0.7 mm MAS rotor, 0.5 mg of material), which corresponds to the maximum MAS frequency achieved to-date, eliminates the need for deuteration to achieve high resolution29, 30. On the other hand, simulations indicate that MAS frequencies on the order of 250 kHz are necessary to yield comparable proton resolution31.
In this work, we compare 1H, 13C correlation spectra obtained for protonated and deuterated samples of a microcrystalline protein at a MAS rotation frequency of 111 kHz, and show that the spectral quality obtained from deuterated samples is superior to those acquired using protonated samples in terms of resolution and sensitivity.
Results
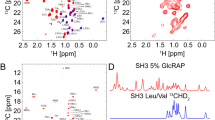
Figure 1 shows the 1H,13C correlation spectra obtained for a protonated and two types of deuterated samples of a microcrystalline sample of the α-spectrin SH3 domain. The 25% and 5% deuterated RAP samples are prepared using u-[2H,13C]-glucose, and a D2O based M9 growth medium that has been diluted with 25% and 5% H2O to yield selective incorporation of protons at the aliphatic sites, respectively24. The SH3 sample in which pro-R or pro-S methyl groups of valine and leucine residues are specifically protonated was produced using α-ketoisovalerate as a precursor for amino acid biosynthesis, yielding CH3 isotopomers)32, 33.

Comparison of MAS solid-state NMR 1H,13C correlation spectra obtained for protonated and deuterated samples of a microcrystalline α-spectrin SH3 domain. Top: Methyl region of the spectra from protonated (left) and α-ketoisovalerate (CH3) labelled (right) samples33 (recorded with B0 = 11.8 T at 111 kHz MAS frequency). Middle: Methyl region of 1H,13C correlation spectra from 25% (left, recorded with B0 = 20 T at 40 kHz MAS frequency) and 5% (right, recorded with B0 = 14.1 T at 20 kHz MAS frequency) RAP24 labelled samples. Bottom: Backbone Hα,Cα correlation spectra for protonated (left) and 25% RAP (right) labelled samples.
We find that the methyl cross peak intensities in the 1H,13C correlation spectra obtained using the protonated sample vary significantly in comparison to the spectra obtained using the α-ketoisovalerate (CH3) labelled sample. A spectrum comprising the full spectral width is provided in the Supporting Information (Fig. S1). In particular, several methyl peaks are broadened or missing in the spectrum from the protonated sample. Using the RAP labelled samples, high resolution Hα,Cα correlation spectra are obtained. The resolution of the spectrum from the 25% RAP labelled sample is only compromised due to the presence of three methyl isotopomers CH3, CH2D, CHD2 which yield isotope induced chemical shift changes34. This problem can be alleviated by using 5% RAP labelled sample. In the protonated sample, the α-carbon spectral region seems to be rather well dispersed at first sight. However, cross peaks significantly overlap in particular in the region of around 4.7/55 ppm proton/carbon chemical shifts.
To quantify the differences between the spectra, we analysed the experimental 1H and 13C line widths and the sensitivity in detail. The results are summarized in Fig. 2. Representative traces for two residues along the proton and carbon dimension of the correlation experiment are shown in Fig. 2A. Comparing the α-ketoisovalerate (CH3) labelled sample and the fully protonated sample, we observe an average gain in resolution on the order of 3 and 2 in the proton and carbon dimensions, respectively (Fig. 2C,D). To account for the difference in the amounts of material in the two rotors (Fig. S2), we quantified the amount of sample by performing 1D-13C experiments using direct 13C excitation. This ratio (~1.76 times) is used to normalize the cross peak intensities represented in Fig. 2B. In total, we find approximately a 2–3x higher sensitivity for the α-ketoisovalerate (CH3) labelled sample. This enhancement could potentially be further increased by an additional factor of 2 by making use of stereospecific precursors for amino acid biosynthesis35.

(A) 1D traces from the 1H,13C correlation spectra represented in Fig. 1 along the 1H and 13C dimension for residues L8 and V44. Spectra from the protonated and the α-ketoisovalerate labelled (CH3) sample are depicted in red and black, respectively. (B) Cross peak intensities for all rigid methyl residues in the protonated (red square) and deuterated (black triangle) α-spectrin SH3 domain. The error bar is within the symbol size. The intensities are normalised according to the sample amount as described in Fig S2. (C,D) 1H and 13C line width for all methyl bearing residues in the α-spectrin SH3 domain for the protonated (red square) and deuterated (black traingle) sample. The error of the line width is given by the acquisition times in the proton (35 ms) and the carbon (70 ms) evolution periods, respectively. The dashed lines represent the average line width for proton and carbon resonances, respectively. All experiments were carried out at B0 = 11.8 T (500 MHz 1H Larmor frequency).
We attribute differences in peak intensities to the effective intra- and inter-methyl proton-proton dipolar couplings in the sample. To estimate the influence of the proton network around a given proton site, we quantified the spin density, following the convention of Zorin et al.36. In solids, a given spin is affected by many internuclear couplings of different magnitudes. Therefore, d RSS, which is defined as the square root of the sum of squared dipolar couplings, provides a good approximation for an effective coupling experienced by a given spin
Figure 3 shows the d RSS values for amide and methyl protons in the α-spectrin SH3 domain (PDB-ID: 2nuz)37. In the calculation, a distance cut-off of 10 Å has been employed.

Effective 1H, 1H dipolar interactions d RSS for amide and methyl protons in the α-spectrin SH3 domain. The protonated and deuterated protein are represented in red and black, respectively. For the calculation of d RSS for amide protons in the deuterated sample, only exchangeable protons are assumed to be present, whereas for the protonated sample, both exchangeable and non-exchangeable protons were taken into account. d RSS has been calculated using a distance cut-off of 10 Å and (Eq. 1), employing the coordinate file 2nuz of the PDB database.
In the deuterated protein sample (back-substituted with 100% protons at exchangeable sites), the average proton, proton dipolar couplings for amides are on the order of 7 kHz, and thus approximately 3x smaller in comparison to the effective dipolar couplings in the protonated sample. In the deuterated protein sample, high d RSS values are obtained primarily in the turn regions (RT-loop around residue 20, the N-src-loop at residue 40, the distal loop with residues 47–49), for which short amide-amide distances are found. Apparently, a MAS rotation frequency of around 50 kHz is sufficient to yield efficient averaging of d RSS for the amide protons in perdeuterated samples. Under these conditions, average 1HN line widths are on the order of 50 Hz14.
For the selectively methyl protonated sample, the effective proton, proton dipolar coupling is on the order of 27 kHz for methyl protons (Fig. 3). This value is similar in magnitude to the Hα d RSS values for a protonated sample (Figure S3). The 1Hα,13Cα correlation spectrum for a protonated sample at a MAS rotation frequency of 111 kHz is reasonably well dispersed with an average proton line width on the order of 100 Hz (Figure S3), suggesting that this spinning frequency is sufficient to yield efficient averaging of d RSS in selectively methyl protonated samples. In contrast, the d RSS values for methyl protons in a fully protonated sample are approximately 1.5x larger, indicating that MAS frequencies on the order of 160 kHz and above are necessary to yield methyl 1H,13C correlation spectra with maximum resolution. Effective dipolar interactions are apparently not efficiently averaged in a protonated sample at a MAS frequency of 110 kHz. This in agreement with a previous study, where it has been suggested that homogeneous interactions require MAS frequencies that are significantly larger (10 times) than the strength of the interaction31.
In order to test whether d RSS is a good descriptor for the proton spin network in MAS solid-state NMR, the calculated effective proton, proton dipolar couplings is represented as a function of the normalized cross peak intensities in Fig. 4. In general, higher Hα peak intensities are found for smaller d RSS values. Proton isotropic chemical shifts are not included in the analysis. n = 0 rotational resonance effects may potentially yield line broadening, which can be alleviated at higher magnetic fields. In a typical protein sample, the effective proton, proton dipolar interactions are on the order of 15–45 kHz. In that sense, rotation frequencies greater than 300 kHz are required in order to efficiently average all homonuclear proton dipolar couplings.

Correlation plot between the effective proton, proton dipolar coupling d RSS and the normalized peak intensities for methyl and Hα protons in a protonated SH3 sample. Experimental data were obtained from experiments that were recorded at a MAS rotation frequency of 111 kHz.
Even though fully protonated samples would be ideal for spectroscopy, a brute-force approach bears risks for structure determination protocols: Resonances of protons, which are involved in strong dipolar interactions, are potentially weak or not observable. These residues do not yield long-range distance restraints (>5 Å). If especially long-range distance restraints are missing, structure calculation protocols might potentially converge into a wrong fold.
Conclusion
We demonstrated that at MAS rotation frequencies of 110 kHz, the spectral quality obtained from selectively methyl protonated samples is superior in comparison to the spectrum obtained from a fully protonated sample. The α-ketoisovalerate labelled (CH3) deuterated sample yields a gain in resolution on the order of 3 and 2 in the proton and carbon dimension, respectively. Sensitivity is enhanced by approximately a factor of 2–3, which could be further increased by a factor of 2 by making use of stereospecific precursors for amino acid biosynthesis. We believe that in particular experiments involving large non-symmetric protein complexes in which sensitivity is limiting will benefit from such deuteration schemes38, 39. The results presented here have been obtained only for a single microcrystalline protein preparation. In comparison to other samples, the α-spectrin SH3 domain yield very narrow line proton spectra. This allows to monitor subtle spectral changes induced by changes in the labelling scheme. We expect that similar effects will be observable for non-crystalline proteins as well, as the physical basis of dipolar interactions is the same, and the average proton density comparable for all proteins.
Methods
The perdeuterated, selectively methyl group protonated microcrystalline SH3 sample was prepared as described previously33. In brief, expression was carried out in 100% D2O M9 medium supplemented with 15N-Ammonium Chloride and D-Glucose-13C, d7. α-ketoisovalerate (2-Keto-3-(methyl-d3)-butyric acid-4-13C, 3-d sodium salt, Sigma Aldrich) was added to the M9 minimal medium 1 h prior to induction with 1 mM IPTG (at OD600 = 0.5–0.6). yielding methyl CH3 isotopomers in a deuterated matrix. Subsequent to overnight expression, the SH3 domain was purified via anion exchange and size exclusion chromatography as described before. For crystallization, pure protein was lyophilized and dissolved in 100% D2O (final concentration: 8–10 mg/ml). Ammonium sulfate (dissolved in 100% D2O) was added to a final concentration of 100 mM and the pH was adjusted to 8.0 by adding NaOD. In order to produce the RAP labelled samples, the deuterated M9 minimal medium was supplemented with H2O (25% H2O and 5% H2O for 25% and 5% RAP samples, respectively)24. The protonated sample was prepared employing only protonated chemicals. The protein samples were not unfolded prior to crystallization, and no paramagnetic dopant was used in the crystallization buffer. The amount of protonation in the samples have been characterised in earlier work18, 40.
The proton detected CP based HSQC experiments for the protonated and α-ketoisovalerate labelled samples were carried out at a static magnetic field of 11.8 T (1H Larmor frequency 500 MHz), employing a 0.7 mm H/C/N MAS probe. The best Hartmann-Hahn condition was found at a nominal RF amplitudes of 70 kHz and 40 kHz on the 1H and 13C channel, respectively. A linear ramp on the 1H channel was applied for the 1H− > 13C CP step (contact time: 800 μs, range of amplitude: 70–100%) and for 13C− > 1H CP (contact time: 500 μs, range of amplitude: 100–70%), respectively. For hard pulses, B1 amplitudes of 156 kHz and 100 kHz were used for 1H and 13C, respectively. Low power decoupling was applied in the evolution periods using swept TPPM during t1 (ω1H/2π = 30 kHz)41, and WALTZ16 (ω13C/2π = 5 kHz) in the direct dimension. Water suppression was achieved using MISSISSIPPI with ω1H/2π = 30 kHz for 300 ms42. Phase-cycling was performed as described in Barbet-Massin et al.17. For all the experiments, the actual sample temperature was calibrated to 288 K using DSS (set temperature: 263 K, VT gas flow rate: 480 L/h). In all experiments, the acquisition times in the F1 and F2 dimensions were set to 70 ms and 35 ms (13C, 1H), respectively. The relaxation delays were set to 0.5 s and 1.5 s in the protonated and α-ketoisovalerate labelled sample, respectively.
The spectrum of the 25% RAP labelled sample was recorded at 20 T (850 MHz) setting the MAS frequency to 40 kHz. The spectrum of the 5% RAP sample was recorded at 14.1 T (600 MHz) with the MAS frequency adjusted to 20 kHz. For experiments involving RAP labelled samples, scalar coupling based HMQC type experiments were employed. 2–3 kHz of WALTZ16 decoupling was used during proton detection.
References
Castellani, F. et al. Structure of a protein determined by solid-state magic-angle spinning NMR. Nature 420, 98–102 (2002).
Wasmer, C. et al. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science 319, 1523–1526 (2008).
Loquet, A. et al. Atomic model of the type III secretion system needle. Nature 486, 276–279, doi:10.1038/nature11079 (2012).
Wang, S. et al. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nature Methods 10, 1007, doi:10.1038/NMETH.2635 (2013).
Mainz, A. et al. The Chaperone αB-Crystallin Deploys Different Interfaces to Capture an Amorphous and an Amyloid Client. Nature Struct. Mol. Biol. 22, 898–905, doi:10.1038/nsmb.3108 (2015).
Wälti, M. A. et al. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl Acad. Sci. USA 113, E4976–4984, doi:10.1073/pnas.1600749113 (2016).
Colvin, M. T. et al. Atomic Resolution Structure of Monomorphic A beta(42) Amyloid Fibrils. J. Am. Chem. Soc. 138, 9663–9674, doi:10.1021/jacs.6b05129 (2016).
Tuttle, M. D. et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nature Struct. Mol. Biol. 23, 409–415, doi:10.1038/nsmb.3194 (2016).
Samoson, A. et al. New horizons for magic-angle spinning NMR. Top. Curr. Chem. 246, 15–31 (2005).
Agarwal, V. et al. De Novo 3D Structure Determination from Sub-milligram Protein Samples by Solid-State 100 kHz MAS NMR Spectroscopy. Angewandt. Chem. Int. Edt. 53, 12253–12256, doi:10.1002/anie.201405730 (2014).
Chevelkov, V., Rehbein, K., Diehl, A. & Reif, B. Ultra-high resolution in proton solid-state NMR at high levels of deuteration. Angew. Chem. Int. Ed. 45, 3878–3881 (2006).
Akbey, Ü. et al. Optimum Levels of Exchangeable Protons in Perdeuterated Proteins for Proton Detection in MAS Solid-State NMR Spectroscopy. J. Biomol. NMR 46, 67–73 (2010).
Reif, B. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: Implications for Structure and Dynamics. J. Magn. Reson. 216, 1–12, doi:10.1016/j.jmr.2011.12.017 (2012).
Lewandowski, J. R. et al. Enhanced Resolution and Coherence Lifetimes in the Solid-State NMR Spectroscopy of Perdeuterated Proteins under Ultrafast Magic-Angle Spinning. J. Phys. Chem. Lett. 2, 2205–2211 (2011).
Knight, M. J. et al. Fast Resonance Assignment and Fold Determination of Human Superoxide Dismutase by High-Resolution Proton-Detected Solid-State MAS NMR Spectroscopy. Angew. Chem. Int. Edt. 50, 11697–11701, doi:10.1002/anie.201106340 (2011).
Penzel, S. et al. Protein resonance assignment at MAS frequencies approaching 100 kHz: a quantitative comparison of J-coupling and dipolar-coupling-based transfer methods. J. Biomol. NMR 63, 165–186, doi:10.1007/s10858-015-9975-y (2015).
Barbet-Massin, E. et al. Rapid Proton-Detected NMR Assignment for Proteins with Fast Magic Angle Spinning. J. Am. Chem. Soc. 136, 12489–12497, doi:10.1021/ja507382j (2014).
Asami, S., Schmieder, P. & Reif, B. High resolution 1H-detected solid-state NMR spectroscopy of protein aliphatic resonances: access to tertiary structure information. Journal of the American Chemical Society 132, 15133–15135, doi:10.1021/ja106170h (2010).
Huber, M. et al. A Proton-Detected 4D Solid-State NMR Experiment for Protein Structure Determination. Chemphyschem 12, 915–918, doi:10.1002/cphc.201100062 (2011).
Wang, S. et al. Nano-Mole Scale Side-Chain Signal Assignment by H-1-Detected Protein Solid-State NMR by Ultra-Fast Magic-Angle Spinning and Stereo-Array Isotope Labeling. Plos One 10, doi:ARTN e0122714, doi:10.1371/journal.pone.0122714 (2015).
Medeiros-Silva, J. et al. H-1-Detected Solid-State NMR Studies of Water-Inaccessible Proteins InVitro and InSitu. Angew Chem Int Edit 55, 13606–13610, doi:10.1002/anie.201606594 (2016).
Sinnige, T., Daniels, M., Baldus, M. & Weingarth, M. Proton Clouds to Measure Long-Range Contacts between Nonexchangeable Side Chain Protons in Solid-State NMR. Journal of the American Chemical Society 136, 4452–4455, doi:10.1021/ja412870m (2014).
Agarwal, V. & Reif, B. Residual Methyl Protonation in Perdeuterated Proteins for Multidimensional Correlation Experiments in MAS solid-state NMR Spectroscopy. J. Magn. Reson. 194, 16–24 (2008).
Asami, S., Schmieder, P. & Reif, B. High resolution 1H-detected solid-state NMR spectroscopy of protein aliphatic resonances: Access to tertiary structure information. J. Am. Chem. Soc. 132, 15133–15135 (2010).
Asami, S., Szekely, K., Schanda, P., Meier, B. H. & Reif, B. Optimal degree of protonation for 1H detection of aliphatic sites in randomly deuterated proteins as a function of the MAS frequency. J. Biomol. NMR 54, 155–168 (2012).
Asami, S. & Reif, B. Assignment strategies for aliphatic protons in the solid-state in randomly protonated proteins. J. Biomol. NMR 52, 31–39, doi:10.1007/s10858-011-9591-4 (2012).
Asami, S. & Reif, B. Proton-detected solid-state NMR at aliphatic sites: Applications to Crystalline Systems. Acc. Chem. Res. 46, 2089–2097 (2013).
Andreas, L. B. et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proceedings of the National Academy of Sciences of the United States of America 113, 9187–9192, doi:10.1073/pnas.1602248113 (2016).
Andreas, L. B. et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc. Natl Acad. Sci. USA 113, 9187–9192, doi:10.1073/pnas.1602248113 (2016).
Stanek, J. et al. NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew. Chem. Int. Ed. Engl. 55, 15503–15509, doi:10.1002/anie.201607084 (2016).
Bockmann, A., Ernst, M. & Meier, B. H. Spinning proteins, the faster, the better? J. Magn. Reson. 253, 71–79, doi:10.1016/j.jmr.2015.01.012 (2015).
Goto, N. K., Gardner, K. H., Mueller, G. A., Willis, R. C. & Kay, L. E. A robust and cost-effective method for the production of Val, Leu, Ile (d1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 13, 369–374 (1999).
Agarwal, V., Xue, Y., Reif, B. & Skrynnikov, N. R. Protein side-chain dynamics as observed by solution- and solid-state NMR: a similarity revealed. J. Am. Chem. Soc. 130, 16611–16621 (2008).
Agarwal, V., Diehl, A., Skrynnikov, N. & Reif, B. High Resolution 1H Detected 1H,13C Correlation Spectra in MAS Solid-State NMR using Deuterated Proteins with Selective 1H,2H Isotopic Labeling of Methyl Groups. J. Am. Chem. Soc. 128, 12620–12621 (2006).
Gans, P. et al. Stereospecific Isotopic Labeling of Methyl Groups for NMR Spectroscopic Studies of High-Molecular-Weight Proteins. Angewandte Chemie Int. Ed. 49, 1958–1962, doi:10.1002/anie.200905660 (2010).
Zorin, V. E., Brown, S. P. & Hodgkinson, P. Origins of linewidth in 1H magic-angle spinning NMR. J. Chem. Phys. 125, 144508 (2006).
Chevelkov, V. et al. Differential Line Broadening in MAS solid-state NMR due to Dynamic Interference. J. Am. Chem. Soc. 129, 10195–10200 (2007).
Barbet-Massin, E., Huang, C.-T., Daebel, V., Hsu, S.-T. D. & Reif, B. Site-Specific Solid-State NMR Studies of “Trigger Factor” in Complex with the Large Ribosomal Subunit 50S. Angewandte Chemie Int. Edt. Engl. 54, 4367–4369, doi:10.1002/anie.201409393 (2015).
Kurauskas, V. et al. Sensitive proton-detected solid-state NMR spectroscopy of large proteins with selective CH3 labelling: application to the 50S ribosome subunit. Chem. Comm. 52, 9558–9561, doi:10.1039/c6cc04484k (2016).
Ollerenshaw, J. E., Tugarinov, V., Skrynnikov, N. R. & Kay, L. E. Comparison of 13CH3, 13CH2D, and 13CHD2 methyl labeling strategies in proteins. J Biomol NMR 33, 25–41, doi:10.1007/s10858-005-2614-2 (2005).
Lewandowski, J. R. et al. Measurement of site-specific 13C spin-lattice relaxation in a crystalline protein. Journal of the American Chemical Society 132, 8252–8254, doi:10.1021/ja102744b (2010).
Zhou, D. H. & Rienstra, C. M. High-performance solvent suppression for proton detected solid-state NMR. J Magn Reson 192, 167–172, doi:10.1016/j.jmr.2008.01.012 (2008).
Acknowledgements
We acknowledge support from the Helmholtz-Gemeinschaft and the Deutsche Forschungsgemeinschaft (Grants Re1435 and SFB-1035, project B07). In addition, we are grateful to the Center for Integrated Protein Science Munich (CIPS-M) for financial support. ZT acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 657682.
Author information
Authors and Affiliations
Contributions
K.X., R.S. performed experiments and analysed data; S.A. provided NMR data on RAP samples; C.M. prepared SH3 samples; DCRC helped in sample preparation; V.D., S.W. performed experiments employing the 0.7 mm MAS probe; K.X., R.S., Z.T., B.R. performed data interpretation, designed research and experimental plans; All authors contributed in writing the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, K., Sarkar, R., Motz, C. et al. Limits of Resolution and Sensitivity of Proton Detected MAS Solid-State NMR Experiments at 111 kHz in Deuterated and Protonated Proteins. Sci Rep 7, 7444 (2017). https://doi.org/10.1038/s41598-017-07253-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07253-1
This article is cited by
-
Site-specific protein methyl deuterium quadrupolar patterns by proton-detected 3D 2H–13C–1H MAS NMR spectroscopy
Journal of Biomolecular NMR (2022)
-
Multi-receiver solid-state NMR using polarization optimized experiments (POE) at ultrafast magic angle spinning
Journal of Biomolecular NMR (2020)
-
Resonance assignment of the outer membrane protein AlkL in lipid bilayers by proton-detected solid-state NMR
Biomolecular NMR Assignments (2020)
-
MAS dependent sensitivity of different isotopomers in selectively methyl protonated protein samples in solid state NMR
Journal of Biomolecular NMR (2019)
-
Alpha protons as NMR probes in deuterated proteins
Journal of Biomolecular NMR (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
