
Abstract
We performed integrative analysis of genes associated with type 2 Diabetes Mellitus (T2DM) associated complications by automated text mining with manual curation and also gene expression analysis from Gene Expression Omnibus. They were analysed for pathogenic or protective role, trends, interaction with risk factors, Gene Ontology enrichment and tissue wise differential expression. The database T2DiACoD houses 650 genes, and 34 microRNAs associated with T2DM complications. Seven genes AGER, TNFRSF11B, CRK, PON1, ADIPOQ, CRP and NOS3 are associated with all 5 complications. Several genes are studied in multiple years in all complications with high proportion in cardiovascular (75.8%) and atherosclerosis (51.3%). T2DM Patients’ skeletal muscle tissues showed high fold change in differentially expressed genes. Among the differentially expressed genes, VEGFA is associated with several complications of T2DM. A few genes ACE2, ADCYAP1, HDAC4, NCF1, NFE2L2, OSM, SMAD1, TGFB1, BDNF, SYVN1, TXNIP, CD36, CYP2J2, NLRP3 with details of protective role are catalogued. Obesity is clearly a dominant risk factor interacting with the genes of T2DM complications followed by inflammation, diet and stress to variable extents. This information emerging from the integrative approach used in this work could benefit further therapeutic approaches. The T2DiACoD is available at www.http://t2diacod.igib.res.in/.
Similar content being viewed by others


Introduction
T2DM is on the rise1. It is characterized by hyperglycaemia due to insulin resistance and decreased insulin activity. Genetic factors, environmental agents and their interactions have been recognized as contributors to the development of T2DM2,3,4. As of 2014, more than 371 million people reportedly suffer from T2DM in several countries with China at the top (92.3 million) followed by India (80 million) and the USA (29.1 million)5. Before the era of genome wide association studies (GWAS), hunting for genetic factors produced a handful of genes such as CAPN10, TCF7L2, PPARG and KCNJ11 6, 7. The association of TCF7L2, PPARG and KCNJ11 with T2DM have been observed in several populations and both PPARG and KCNJ11 proteins are targets of currently used T2DM drugs7. The GWAS era including meta-analysis enabled uncovering of numerous genes and their variants associated with T2DM. Yet these variants apparently are able to explain only about 20–30% of heritable component8. Re-sequencing is now emerging to enable comprehensive variants discovery with its identified potential not offered by imputation and chip based fine-mapping approaches8. These efforts can get further challenged by gene environment interactions whereby phenotypic effects for given genotypes likely vary9. Diet and physical activity are most considered among environmental factors. Dietary components could affect directly (through interaction with targets) and indirectly (through interaction with gut microbiome). Although some compounds such as vitamins and organic pollutants such as heptachlor epoxide and polychlorinated biphenyls have been studied, this area is relatively under investigated and most environmental agents are not known5, 10. Despite these deficiencies the huge body of studies carried out so far including those in animal models have endowed us with many genes associated with T2DM and its associated complications that can serve for identifying drug targets, potential biomarkers and clinical applications.
Several GWAS have been carried out to identify genetic susceptibility to T2DM in different populations11,12,13,14. Recently Fuchsberger et al. analysed the T2DM associated variants by increasing the sample size and observed that most of the variants were located within the same regions previously identified by GWAS14. Studies including meta-analysis enabled identification of 44 genetic loci associated independently with transcriptome data sets of human tissues from T2DM patients. These studies are valuable for gaining insights into the probable pathways leading to T2DM15, 16. Integration of genes co-expression and interaction networks enabled identification of TGFBRII, MAPK, PTPN1, EGFR, and CAV1 pathways, which could lead to cardiovascular and kidney related complications and diabetic nephropathy and Zelezniak et al. integrated skeletal muscle gene expression datasets with human metabolic network reconstructions, which enabled them to identify transcription factors CREB, NRF1 and PPAR family regulatory network connecting several parts of metabolism17, 18. Zhong et al. (2010)19 identified a functionally related set of diabetes susceptibility genes using expression SNPs (eSNPs) comprising an adipose sub-network and subsequently validated malic enzyme (ME1), which converts malate to pyruvate19.
Hyperglycaemic conditions in late stages of diabetic individuals include well known microvascular complications (nephropathy, neuropathy, retinopathy) and macrovascular complications (atherosclerosis and cardiovascular)20,21,22,23. Even these complications evidently are polygenic disorders24,25,26,27,28,29,30. Inflammation could lead to vascular calcification in association with atherosclerosis31. Hepatokines such as Fibroblast growth factor 21, Fetuin A and selenoprotein P are also implicated in the development of atherosclerosis32. It is evident that the complications arising from T2DM are also multifactorial and could be further governed by gene-environment interactions.
We have mined the voluminous literature available in the PubMed database for genes associated with T2DM and other complications. The available specific databases, namely, Type 2 diabetes Database (T2D-Db)33, T2DM Genetic Association Database (T2DGADB)34 and T-HOD35 although serving as useful resources by providing genetic association studies as well as integrated resources including gene expression, pathways and protein–protein interactions, however lack recent updates on the roles of specific genes identified as associated with complications in T2DM. We have supplemented our resource collection with data in these databases in a few cases. We believe, our up to date integrative data collection from genetic and functional gene expression studies would serve better for developing methods for risk assessment of T2DM associated complications and health forecasting.
Results
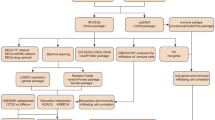
T2DiACoD contains a total of 650 non-redundant genes reported to be directly or indirectly associated with complications under diabetic conditions namely atherosclerosis, diabetic nephropathy, diabetic neuropathy, diabetic retinopathy, cardiovascular. The work flow for text analytics of literature from PubMed is shown in Fig. 1. Among the complications in T2DM condition, nephropathy (403 genes) tops the list followed by cardiovascular (172 genes), retinopathy (161 genes), neuropathy (130 genes), and atherosclerosis (115 genes). 34 microRNAs were associated with these complications and the miRNA hsa-miR-103/107 is common to atherosclerosis, nephropathy and neuropathy complications. Seven genes AGER (advanced glycosylation end product-specific receptor), TNFRSF11B (tumor necrosis factor receptor superfamily, member 11b), CRK (v-crk sarcoma virus CT10 oncogene homolog (avian)), PON1 (paraoxonase 1), ADIPOQ (adiponectin, C1Q and collagen domain containing), CRP (C-reactive protein, pentraxin-related) and NOS3 (nitric oxide synthase 3 (endothelial cell)) are associated with all 5 complications (Fig. 2). The visualization of T2DiACoD is shown in Fig. 3 and structure of database is shown in Fig. 4.

Work flow for text analytics of literature from PubMed. The data sources for each task are mentioned adjacent to the task name.

Venn diagram of genes implicated in T2DM complications. 7 genes encircled are PON1, TNFRSF11B, CRP, NOS3, CRK, AGER, ADIPOQ common in all complications.

Visualization of T2DiACoD. The four sections of T2DiACoD are shown here as cropped screen shots. In T2DM complication, the nephropathy gene is displayed with gene summary (bottom right) and in the population section, example data for each of the populations in the database is shown. Histograms (top left) represent the differential expression of a gene IL6. miRNAs with their corresponding genes and observations are displayed top middle. An example of combination search is presented - nephropathy with diet and stress.

Database structure. T2DiACoD is a tetrapodic layout (i) T2DM associated complications, (ii) Gene expression (iii) miRNA for T2DM (iv) Population studies.
Comparison with other available resources
We have compared T2DiACoD from available resources, namely, DisGeNET36, T2D-Db37 and T2D Knowledge portal38. DisGeNET integrates the data on human-disease association from available databases and literature. Its data are categorized into 3 categories 1) Curated Data 2) Predicted Data and 3) Literature Data. DisGeNET has collected data from different sources i.e. UniProt, ClinVar, Orphanet, The GWAS catalog, CTD, RGD, MGD and literature data from GAD, LHGDN and BeFree. We observed that DisGeNET contains 705 genes, however several of these genes are either associated with Type 1 Diabetes or are false matches (either absence of strong evidence or presence of negative evidence in the text provided). T2D-Db provides information at molecular level for type 2 diabetes and its pathogenesis and there are 83 genes for T2DM associated complications, namely, nephropathy, neuropathy, retinopathy and cardiovascular diseases. T2D Knowledge portal is a database of DNA sequence, functional and epigenomic information, and clinical data from studies on type 2 diabetes and complications as well as provides analysis of this data. T2DiACoD containing 650 genes as on date is a repository, which stores information on genes associated with T2DM complications, protective genes and their roles, Drug targets, population SNPs data, expression analysis in different tissues of T2DM patients, miRNAs, 34 miRNAs and their target genes involved in associated complications of T2DM, interactions with risk factors, namely obesity, inflammation, diet and stress of genes associated with T2DM complications.
Gene trends
We sought to identify the trends of genes studied in each complication by classifying them either as studied in a single year or are being studied in multiple years perhaps even to this day. These trends could be considered for new drug targets or markers for T2DM associated complications. Trends were computed using the Buzz Word Index (BWI). The results are displayed in Table 1 and the detailed results are presented in Supplementary Table 1. A few genes did not register a positive BWI value. Among the 23 drug targets (listed in T2DiACoD web site), 13 (SGK1, SLC2A4, JAK2, HPSE, GCK, DPP4, CCR2, GIP, APOC3, PCSK9, PTPN1, SLC5A2, VCAM1) have been studied in multiple years, three (GLP1R, ERRFI1, LRP2) have been studied in a single year. In case of markers or biomarkers, 2 genes each were studied in multiple (CXCL16, RBP4) as well as in a single year (FGF21, HLA-DQA1). Below, we describe the highlights of the results of trends of genes in each T2DM complication (Supplementary Table 6 for acronyms expansion).
Cardiovascular
In the case of cardiovascular disease, out of 172 genes there are 31 genes studied in a single year. The gene ACVR1C (activin A receptor type 1C) studied in 2015 with high BWI value 1547.5 is a member of TGF-beta receptors and is reportedly correlated in expression with pathogenic risk of T2DM as well as cardiovascular diseases. Silencing of ACVR1C/ALK7 gene has reportedly protective effect on diabetes induced aortic stiffness, insulin resistance and hyperlipidemia39. 129 genes were studied in multiple years including PON1, NOS3, TNF (tumor necrosis factor), TNFRSF11B (OPG), HMGB1 (high mobility group box 1), and ADIPOQ. The gene PON1 (paraoxonase 1), an anti-inflammatory high density lipoprotein (HDL)-associated enzyme, has been shown to decrease in diabetic patients40, 41, and its low concentration and enzymatic activity could be independent predictor of cardiovascular events in diabetic patients42. The synergic effect of NOS3 (eNOS) Asp298 allele confirms its pathological role for cardiovascular diseases in T2DM patients43. Blazquez-Medela et al. (2012) studied relationship between serum TNFRSF11B (Osteoprotegerin (OPG)) and vascular alteration in associated pathologies and its levels were found to be higher in diabetic, hypertensive with retinopathy and cardiovascular risk patients and therefore associated with cardiovascular risk in diabetes and hypertension patients44. The G allele of ADIPOQ (adiponectin) G276T polymorphism is a susceptible allele for cardiovascular disease in T2DM patients45 and increased serum HMGB1 level was associated with cardiovascular diseases in T2DM patients46.
Atherosclerosis
28 Genes were studied in a single year in atherosclerosis. DHCR7 (7-dehydrocholesterol reductase) with BWI 135.8 and B2M (beta-2-microglobulin) with BWI 359.8 had high BWI values indicating good emphasis of the corresponding genes in these studies. DHCR7 was recently investigated for its role in T2DM to influence subclinical atherosclerosis using Mendelian randomization approach and carotid artery intima-media thickness (cIMT) measurements47. Strawbridge et al. observed rs3829251 (DHCR7) influenced the progression of atherosclerosis in T2DM patients. Glycation of B2M may contribute to the risk of diabetes associated complications. Kim et al. in 2014 reported higher serum B2M as an independent risk factor for atherosclerosis and diabetic nephropathy (DN) in T2DM patients48. Genes IRAK4, HDAC9, ACSL1 and APPL159 are recently reported as atherosclerosis causing genes in T2DM patients. 59 genes were reportedly being investigated in multiple years for their role in atherosclerosis, for example the genes ABCA1, PON1, MTHFR, PLTP, PPAR, TNFRSF11B (OPG), FABP4, and IL18 are being studied for several years since 1997 with varying emphasis. ABCA1 (ATP-binding cassette transporter A1) was noted as atheroprotective protein49 in that it protects against the formation of atherosclerosis and macrophage ABCA1 protects arteries from promoting atherosclerosis lesions50. Polymorphism or genetic aberrations in ABCA1 gene can be associated with severity of atherosclerosis51. Sartippour et al. (2000) evaluated the in vitro and ex vivo effect of high glucose concentrations on macrophage PPAR (Peroxisome proliferator-activated receptor) mRNA expression52 and observed that dysregulation of macrophage PPAR expression in T2DM alters arterial lipid metabolism and inflammatory response and might contribute to the accelerated atherosclerosis in T2DM. Upregulation of FABP4 (fatty acid binding protein 4) further enhances the macrophage lipid accumulation by advanced glycation end products (AGE), which further accelerates formation of foam cells and development of atherosclerosis in diabetic patients53. Nakamura et al. studied whether serum level of IL-18 (Interleukin 18) is a common predictor of nephropathy and atherosclerosis in T2DM patients. They observed that serum and urinary level of IL-18 were significantly elevated in T2DM patients compared to controls thereby indicating that serum levels of IL-18 could be a predictor for these complications54.
Nephropathy
Diabetic nephropathy is a widely studied complication and the list of genes reportedly involved in the cause or progression of diabetic nephropathy is larger than in other complications. There are 127 genes out of 403 studied in a single year. A few examples are, CHN2, CNR, GAS1, IL1B, IRS2, PFKFB2, RREB1, and TRIB3. Genes LRP2, NGAL, IGFBP7, CUBN, CHIT1 are recently reported genes in diabetic nephropathy. The TRIB3 ‘G’ allele was identified as associated with diabetic nephropathy and it was suggested that this observation may help to improve targeting of therapy to diabetic patients55. Zhang et al. characterized diabetic nephropathy (DN) by mesangial proliferation and glomerular hypertrophy. microRNAs are implicated in this pathogenesis. They discovered hsa-miR-34a regulated mesangial proliferation and glomerular hypertophy by directly inhibiting GAS1 in early DN56. In case of IL1B the C511T variant is reported associated with development of DN57. IRS2 is discussed as marker or mediator of human DN58. RREB1 is considered as novel candidate gene for T2DM associated kidney diseases59. 163 genes out of 403 are studied in multiple years. The genes for example, AGT, CTGF, EPO and MTHFR were observed as being investigated with importance. The C677T mutation in MTHFR (Methylenetetrahydrofolate reductase) gene was reported as risk factor for DN in 1999 by Shcherbak et al.60 and Mazza et al.61 and was highly studied gene in 1999 with BWI 436.8.
Neuropathy
In diabetic neuropathy, 31 genes were studied in a single year, for example, HMGB1, IGFBP5 and SERPINF1 (PEDF). Abu El Asrar et al. (2014) hypothesized that increased expression of HMGB1 (high mobility group box-1) gene, a proinflammatory cytokine, is responsible for pathogenic role in mediating diabetes induced retinal neuropathy and observed that early retinal neuropathy of diabetes involves up-regulated expression of HMGB1 and can be mitigated by inhibition of HMGB1 62. Simon et al. (2015) observed that elevated expression of IGFBP5 in diabetic nerves of mice leads to the progressive neurodegeneration and could offer novel treatment strategies for diabetic neuropathy (DNP)63.
The number of genes studied in multiple years are 33 including ACE and NGF. Hellweg et al. (1990) suggested that NGF is required for the development and maintenance of peripheral neurons, therefore changes in their levels could underlie diabetic neuropathy64. Fradji et al. (2013) measured the serum levels of NGF and found lower level of NGF in patients’ with diabetic neuropathy65. Recently in 2014 it was observed that Vitamin A increases the level of NGF, which helps in improving diabetic neuropathy in rats66.
Retinopathy
In diabetic retinopathy 34 genes including AP15, GRB2, IL17A, PLXDC1, C5 and L1CAM were studied in a single year in which, C5 and L1CAM are reported as recently as in 2016. For example, Burdon et al. (2015) reported genetic variation on chromosome 17q25.1 near GRB2 as associated with diabetic retinopathy and expression of GRB2 is up-regulated during retinal stress and neovascularisation67. Yamaji et al. (2008) determined whether TEM7 (PLXDC1) is associated with formation of fibrovascular membranes (FVMs) further responsible to cause proliferative diabetic retinopathy (PDR) and observed significant role of TEM7 in the proliferation and maintenance of neovascular endothelial cells in FVMs and also could be molecular target for new diagnostic of PDR68.
Nearly 48.44% of genes (78 genes) were studied in multiple years including ACE, AGER, ADIPOQ, SERPINF1 and SIRT1. Pradeepa et al. (2015) recently proposed to assess the association of ADIPOQ (adiponectin) and microvascular complications of diabetes and observed that serum adiponectin levels were associated with diabetes associated complications and also with severity of retinopathy69. Niu et al. (2012) provided convincing evidence that RAGE (AGER) gene 1704T allele is associated with increased risk of diabetic retinopathy70.
Gene enrichment analysis
The numbers of genes annotated with few gene ontologies (GOs) are far greater than that annotated with a large number of GOs in all 5 complications.
The GOs unique to cardiovascular complications are: circulatory system process, blood circulation, lipid localization, lipid transport, lipid binding. The cardiovascular complications arise due to misbalanced lipid levels in the blood and are considered to be responsible to affect blood vessels; Coronary heart disease (CHD) occurs when arteries supplying blood to the heart muscle becomes blocked. Therefore, blood circulation and lipid transport significantly affect cardiovascular functions. The genes ADIPOQ, ACE2, APOE and CETP are implicated in these processes and polymorphism in these genes have been reported responsible to cause cardiovascular diseases (CVD) in diabetic patients71,72,73,74. The unique GOs found for genes implicated in cardiovascular disease are displayed in Supplementary Table 2a.
In the case of atherosclerosis 27 unique GOs were observed, namely, lipid rich processes, cholestrol homestasis, lipoprotien metabolc process, high density particle process. Ait-Oufella et al. (2011)75 reviewed the role of cytokines in the cause of atherosclerosis and observed that blockage of proinflammatory cytokines could limit the plaque development and progression75. Genes implicated in these GOs are for example IL6, TNF, TLR4 and ADIPOQ. It is reported that homocysteine upregulates Matrix metalloproteinases-tissue inhibitor of metalloproteinases (MMP-TIMP) pathway and IL6 release, the effect being stronger in the presence of high glucose, which further contributes to increased atherogenesis in diabetic patients76. Advanced glycation end-product of low-density-lipoprotein (AGE-LDL) activates TLR4-mediated signaling pathway, thus inducing proinflammatory cytokine production, with increased risk of atherosclerosis in diabetics77. The unique GOs found for genes implicated in atherosclerosis are displayed in Supplementary Table 2b. The GO enrichment map displayed in Fig. 5 shows the high similarity between the two maps of atherosclerosis and cardiovascular.

Gene-set enrichment visualization using EnrichmentMap. In the graph, nodes represent gene set involved in the GO term mentioned adjacent to the node and edges represent the extent of overlap of genes between the two gene sets connected by them. Node size and edge width are proportional to the number of genes. Colour representation Purple = Extracellular space, Violet = Extracellular region, Orange = Positive regulation of peptidyl-tyrosine phophorylation, Olive = Chemokine activity, Army green = Hormone activity, Pine green = Immune response, Light blue = Growth factor activity, Turquoise = Low-density lipoprotein particle binding, Yellow green = Lipid binding, Pink = Lipoprotein metabolic process.
In micro-vascular complications, large numbers of unique GOs are enriched, for e.g. 47 in nephropathy, 25 in retinopathy and 20 in neuropathy. Diabetic nephropathy (DN) is accompanied by renal failure78, 79 and is associated with cardiovascular complications80. Ni et al. discussed the various signaling pathways with respect to development, progression and prevention of hyperglycaemia induced diabetic nephropathy81. It is notable that the number of genes implicated in DN far exceeds than that in other diabetic complications. Consequently, we observed higher number of unique GOs (41) associated with the genes implicated in DN. Regulatory processes, signaling, kinase, redox coupling encompasses diabetic nephropathy. the genes in MAPK and JAK-STAT pathways are associated with these GOs and involved in the pathogenesis of DN82. Overall, the GO terms unique for the genes implicated in DN explain its pathophysiology and therefore these genes could specifically be investigated for their potential for marker. The unique GOs found for genes implicated in diabetic nephropathy are displayed in Supplementary Table 2c.
Diabetic retinopathy is the result of damaged blood vessels supplying blood to retina due to high sugar level. The unique GOs dominant in regulation. Genes such as CTGF, VEGFA, VEGFC and TCF7L2 are implicated in these processes. TCF7L2 promotes pathological retinal neovascularization via ER stress-dependent up regulation of VEGFA 83. CTGF (connective tissue growth factor), downstream effectors of angiogenesis in diabetic retina, could be possible target for therapeutic application of diabetic retinopathy84. Kaidonis et al. (2015) investigated the association between SNP in VEGFC and diabetic retinopathy (DR) in T1DM and T2DM patients’ and observed significant associations, namely, three VEGFC SNPs associated with DR: rs17697419, rs17697515 and rs233342685. In addition we found ALDH1A1, RBP4, APOB, APOA1, RBP1, APOE and RHO genes enriched in retinoid metabolic process as unique GO in diabetic retinopathy. The unique GOs found for genes implicated in diabetic retinopathy are displayed in Supplementary Table 2d.
Diabetic neuropathy is associated with capillary dysfunction86, which is clinically manifested by vascular and metabolic alterations87, 88. The unique GOs encompass hormone activity, regulation and immune response. Tsuzuki et al. (1998) determined the influence of APOE phenotype in the progression of peripheral neuropathy in diabetics89. The decreased expression of SOD2 (superoxide dismutase) increases the risk of diabetic neuropathy90. PDE5 (phosphodiesterase-5) is upregulated in diabetic condition, its inhibitor sildenafil activates cGMP/PKG signaling pathway and mediates beneficial effect on diabetic neuropathy91. The unique GOs found for genes implicated in diabetic neuropathy are displayed in Supplementary Table 2e. The GO enrichment map displayed in Fig. 5 shows the distinctness of nephropathy map but high similarity between the two maps of neuropathy and retinopathy.
miRNAs
34 miRNAs and their target genes involved in associated complications of T2DM are shown in Supplementary Table 3. The target for hsa-miR-103/107 common to atherosclerosis, nephropathy and neuropathy complications is CAV1 involved in viral myocarditis, Endocytosis, Proteoglycans in cancer, Focal adhesion and Bacterial invasion of epithelial cells pathway.
Significant accumulation of malonyl CoA accompanied by ER stress induction is mediated by over-expression of miR-107. Increase in levels of miR-107 is critical and promotes lipid accumulation in hepatocytes and this might form the basis of diverse etiologies encountered in a fatty liver. Lipid accumulation is induced by miR-107 and this is mediated by endoplasmic reticulum (ER) stress. The ER stress inhibitor, 4-Phenyl butyric acid (4-PBA) significantly decreased such miR-107 induced lipid accumulation. db/db mice are well-known genetic models for NAFLD. hsa-miR-107 levels that are elevated in the db/db mice liver, induce ER stress and promote lipid accumulation in liver cells by targeting Fatty acid synthase (FASN)92.
Two members of hsa-miR-33 family called mir-33a and mir-33b, are located in intronic regions within two protein-coding genes for Sterol regulatory element-binding proteins (SREBP-2 and SREBP-1) respectively. It has been shown that adenoviral hsa-miR-33a overexpression in human or mouse islets reduced ABCA1 expression, decreased glucose-stimulated insulin secretion, and increased cholesterol levels. Therefore, hsa-miR-33a regulates ABCA1 expression in pancreatic islets, thus affecting cholesterol accumulation and insulin secretion93.
Two miRNAs hsa-miR-192 and hsa-miR-193b are increased significantly in the pre-diabetic state. Strikingly, in plasma of glucose-intolerant mice these miRNAs are also increased. After a therapeutic intervention consisting of chronic exercise, the circulating levels of hsa-miR-192 and hsa-miR-193b returned to baseline in both pre-diabetic humans and glucose-intolerant mice thereby succeeding in normalizing metabolic parameters94.
Hsa-miR-194 is under regulation by HNF1A and a higher expression level is found in liver and intestinal epithelial cells. HNF1α is important for proper functioning of β-cell and mutations in this gene causes maturity onset diabetes of the young (MODY)95.
Obesity-induced overexpression of hsa-miR-802 impairs glucose metabolism through silencing of HNF1B 96. The levels of circulating hsa-miR-101, hsa-miR-375 and hsa-miR-802 are significantly increased in T2DM patients versus non-glucose tolerance (NGT) subjects and they may become the new biomarkers for type 2 diabetes97.
microRNA 21 encoded by the MIR21 gene was one of the first mammalian microRNAs identified. The dynamic biomarkers for systemic inflammatory or angiogenic status are circulating miR-21-5p and miR-126-3p. The expression levels in circulating angiogenic cells (CACs) from T2DM with major cardiovascular events (MACE) suggest a shift from a proangiogenic to a proinflammatory profile98. The hsa-miR-21 modulates the PTEN-AKT pathway and thus antagonises the insulin resistance in adipocytes. The hsa-miRNA-21can be a new therapeutic target for metabolic disease such as T2DM and obesity99.
The hsa-miR-181 microRNA precursor is a small non-coding RNA molecule regulating SIRT1 and improves hepatic insulin sensitivity. Inhibition of miR-181a might be a potential new strategy for treating insulin resistance and T2DM100.
The level of beta-cell apoptosis increases with small changes in hsa-mir-34a. Further studies can be carried out for determining the effect of rare variants on type 2 diabetes101.
Encoded by the MIR155 host gene, hsa-miR-155 in humans plays an important role in various physiological and pathological processes. Exogenous molecular control in vivo of miR-155 expression may inhibit malignant growth, viral infections, and attenuate the progression of cardiovascular diseases. Down regulated levels of hsa-miR-155 could play an important role in the pathogenesis of T2DM due to their relationship with metabolic control102. Hsa-miR-155 has a direct target NR1H3 (LXRα), which is potentially responsible for liver phenotype of miR-155(−/−) mice. This micro RNA also plays a crucial role in regulation of lipid metabolism as its dysregulation might lead to hepatic steatosis in diabetic patients103.
Silencing of the most abundant miRNA, hsa-miR-124a expressed in neuronal cells in T2DM islets results in increased expression of target genes important for beta cell function. Simultaneously, overexpression of these genes stimulates insulin secretion indicating that expression of miR-124a might contribute to beta cell dysfunction in T2DM104.
Transcription of a “common miRNA signature”, hsa-miR-144 precursor is activated by GATA4. Signature miRNAs, that could possibly explain the pathogenesis of T2DM and the significance of the miRNA in insulin signalling105.
Insulin receptor Substrate 1 (IRS1) a target for hsa-miR-144 and is involved in insulin signaling at mRNA and protein level is also directly down-regulated with increased expression of the miRNA. Plasma miR-144 is increased in patients with T2DM and has also been reported to be involved in regulation of insulin sensitivity in muscle tissue106.
Peroxisome proliferator-activated receptor gamma co activator (PGC)-1α and estrogen-related receptor (ERR)-α expression were upregulated by both overexpression and downregulation of hsa-mir-106b. hsa-miR-106b targets MFN2 and regulates skeletal muscle mitochondrial function and insulin sensitivity. This could offer a potential for new strategy for treating insulin resistance and sensitivity and T2DM107. Brown adipose tissues of high fat diet-induced mice have higher expression levels of hsa-miR-106b and hsa-miR-93. Moreover, the mRNA level of UCP1 was suppressed by the expression of the two miRNAs, hsa- miR-106b and miR-93108.
The two miRNAs (hsa-miR-25 and hsa-miR-92a) could control insulin synthesis directly by targeting the 3′UTR of insulin gene (INS). Furthermore, introduction of anti-miR-25 or hsa- miR-92a leads to increased insulin synthesis, which was suppressed by miR-9109.
MicroRNA (miRNA)-463-3p direct targets ABCG4 inhibits Glucose-stimulated insulin secretion (GSIS) essential for control of metabolic fuel homeostasis110. In type 2 diabetes human pancreatic islets, as compared with non-diabetic controls, up - regulation of insulin and expression of miRNA-463-3p and down-regulation of ABCG4 were observed and their expression levels were closely related.
miR-194 involved in PI3K-Akt signaling pathway was significantly reduced by 25% to 50% in both rat model and human with pre-diabetes and established diabetes111. hsa-miRNA-194 targets Akt1. Interestingly, hsa-miR-194 was a unique miRNA that appeared regulated across different stages of the disease progression from the early stages of insulin resistance to the development of T2DM111.
hsa-miRNA-15b having targets INSR and CCND1 was disregulated by diet-induced obesity (DIO) or palmitate in hepatocytes112. Mice with diet-induced Obesity developed hyperglycemia and insulin resistance associated with a reduction in expression of Insulin receptor (INSR). In addition, protein expression of INSR was suppressed by the over expression of hsa-miR-15b through targeting INSR 3′ untranslated region directly112. This resulted in an impairment of insulin signaling and glycogenesis in hepatocytes. A causal link of miR-15b has been reported to the pathogenesis of hepatic insulin resistance in SFA-induced obesity. INSR gene is involved in 12 pathways of which, Type II diabetes mellitus, PI3K-Akt signaling pathway, Insulin signaling pathway (FDR < 0.05) are prominent related to Type 2 Diabetes whereas CCND1 is involved in Jak-STAT signalling pathway apart from PI3K-Akt signaling pathway112.
A significant increase of hsa-miR-10a was observed with the knockdown of HDAC3 with siRNA, resulting in decrease of CREB1 the target of miR-10a, and fibronectin (FN) expression in kidney of HFD/STZ mice113. On the contrary, over expression of HDAC3 decreased miR-10a content, enhanced albumin-to-creatinine (ACR) value, CREB1 and FN in mice. To sum up,it has been elucidated that HDAC3/miR-10a/CREB1 serves a new method underlying kidney injury,providing potential therapeutic target in type 2 diabetes113.
Protective Genes
During the search for genes responsible for T2DM associated complications we observed some genes could offer protection from the disease either by their up regulation or down regulation or genetic association. The numbers of genes offering protective effect were 33 in nephropathy, 14 in neuropathy, 13 in retinopathy, 11 in cardiovascular diseases and 4 in atherosclerosis. We observed 19 of these genes differentially expressed in T2DM patients with Padj <=0.05, namely, ADCYAP1, ALDH2, APPL1, CAT, CDKAL1, DCXR, ERBB3, FGF21, HDAC4, HLA-DQA1, NFE2L2, NGF, PECAM1, PROC, RBP4, SIRT6, SYVN1, TIMMP4 and TRPV4. None of the genes had fold change (FC) either >2 or <0.5. Example genes with function and mechanism of protection against respective complications are described below.
A few genes ACE2, ADCYAP1, NCF1, NFE2L2, OSM, SMAD1, TGFB1, BDNF, SYVN1, TXNIP, CD36, CYP2J2 and NLRP3 are described below with their function and protective role in corresponding diseases.
Cardiovascular
The NLRP3 and CYP2J2 provide protection against cardiovascular (CVD) complications in T2DM patients. NLRP3, NLR family, pyrin domain containing 3, is a member of NALP3 inflammasome complex. Silencing the NLPR3 gene might protect against diabetic cardiomyopathy114. CYP2J2, cytochrome P450, family 2, subfamily J, polypeptide 2, is a member of cytochrome P450 superfamily of enzymes and is responsible for epoxidation of endogenous arachidonic acid in cardiac tissues. Cardiac specific overexpression of CYP2J2 protect against diabetic cardiomyopathy115.
Atherosclerosis
CD36 is a lipid and fatty acid receptor that plays an important role in the metabolic syndrome and associated cardiac events116. In rodent models Geleon A et al. show that CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis.
The gene NFE2L2 contributes in the protection against all 3 micro-vascular complications. ACE2 and ADCYP1 offer protection against diabetic retinopathy as well as diabetic nephropathy. NCF1, OSM, SMAD1 and TGFB1 provide protection against diabetic nephropathy, SYVM1 and TXNIP protect against diabetic retinopathy and BDNF in ameliorating the diabetic neuropathy.
The gene NFE2L2, Nuclear factor, Erythroid 2 Like 2, is a transcription factor, regulating oxidative stress and also has an anti-inflammatory effect. In nephropathy, activation of NFE2L2 reduces oxidative damage and negatively regulates TGFB1 & extracellular matrix production117. In retinopathy, Xu Z et al. has described the protective role of NFE2L2 in retina. NFE2L2 regulates antioxidant genes via binding of ARE (Antioxidant Response Elements) and NFE2L2/ARE dependent signalling can cancel out diabetic retinopathy mediated injuries in retinal neurons118. In Neuropathy the expression of NFE2L2 & HMOX1 is down-regulated in sciatic nerves of diabetic mice and its expression aids in reduction of formalin induced inflammatory pain and thereby indicating its role in preventing sensory motor alterations118. Negi et al. found that decrease in NF-κB activation cascade and oxidative stress by increasing NFE2L2 may offer neuro-protective effect in diabetic neuropathy119.
ACE2, angiotensin 1 converting enzyme 2, functions in the regulation of cardiovascular and renal functions. Its expression is gradually decreased resulting in accumulation of ang11 in kidney leading to renal injury120. Increased expression of ACE2 overcomes the impaired balance of retinal RAS and confers protection against DR121. ADCYAP1, adenylate cyclase activating polypeptide 1, stimulates adenylate cyclase and cyclic adenosine monophosphate (cAMP) levels. ADCYAP1 provides protection in diabetic retina by attenuating neuronal cell loss in DR and mediating through the activation of PAC-1 receptor122. Its anti-inflammatory, anti-apoptotic and anti-fibrotic properties could aid in ameliorating DN.
Nephropathy
NFE2L2, nuclear factor, erythroid 2, is a multicomponent enzyme activated to produce superoxide anions. It provides protection against DN through the inhibition of TGF-beta1 and reduction in the production of extracellular matrix123. OSM, oncostatin M, is a member of cytokine family and regulates production of cytokines IL-6, G-CSF and GM-CSF. Tubular epithelial cell-myofibroblast transdifferentiation (TEMT) induced by OSM by activating JAK/STAT pathway could be inhibited by SOCS. Liu et al. reported that SOCS proteins inhibit OSM as well as TEMT induction and has a therapeutic effect in DN124. SMAD1, SMAD family member 1, is a signal transducer and transcriptional modulator. Decrease in SMAD1 and collagen type IV provides protection in DN22. TGFB1, transforming growth factor, beta 1, is a multifunctional peptide that regulates proliferation, differentiation and adhesion. Increased TGFB1 increases the production of extracellular matrix space (ECM) through activation of NOX4. Reduction in NOX4/TGFβ-1 signaling may provide therapeutic potential against DN125.
Neuropathy
In neuropathy, BDNF, brain derived neurotrophic factor, is a member if nerve growth factor family. It is necessary for the survival of striatal neurons in the brain. Increased BDNF likely contributes to reduction in Kv channel function through TrkB receptor stimulation with potential therapeutic effects in diabetic neuropathy126.
Retinopathy
In retinopathy, SYVN1, syniviolin 1, is involved in endoplasmic reticulum associated degradation. SYVN1 confers diabetic retinopathy resistance. Yang S et al. described this with expression analysis and found lower expression of SYVN1 in diabetic mice127. TXNIP, thioredoxin interacting protein, is a thioloxidoreductase. It protects cells from oxidative stress. TXNIP plays crucial role in the inflammation and retinal injuries in early stages of DR128.
Drug Targets
We have collected data on 23 recently identified drug targets for T2DM and its associated complications. 16 drug targets are reported for T2DM only and 7 others are for associated complications. For e.g. increased expression of solute carrier family 2, member 4 (SLC2A4) is beneficial for the treatment of insulin resistance129. Only JAK2 was differentially expressed and down regulated in T2DM patients’ adipose tissue. JAK2 is associated with diabetic nephropathy. JAK2 is recognized target for T2DM targeting JAK-STAT pathway130.
Differentially expressed genes from population studies
The genes collected from GWAS study in different populations namely Indian, American, Chinese, Japanese, European and Mexican, were examined for their differential expression in T2DM patients. These differentially expressed genes could possibly underlie fundamental biological processes perhaps beyond genetic differences between individuals. Out of 500 genes reported from all populations where 249 are unique genes, we obtained 29 differentially expressed genes and 2 genes had FC either >2 or <0.5. These genes are IRAK1 and VEGFA. VEGFA is up regulated (FC = 7.91) in skeletal muscle tissues of T2DM patients. VEGFA is associated with 4 complications: nephropathy, neuropathy, retinopathy and atherosclerosis131,132,133. IRAK1 is down regulated (FC = 0.008) in skeletal muscle tissues of T2DM patients and it is associated with neuropathy.
Differential expression, multiple tissue involvement, presence in plasma and risk factors
We analysed individually 11 datasets published on T2DM patients’ tissues pancreas, adipose, skeletal muscle and liver. We have considered differentially expressed genes in different tissues in any of the microarrays, because there could be multiple pathways leading to the disease. We obtained a total of 227 differentially expressed T2DiACoD genes. 191 genes were differentially expressed in pancreas, 34 genes were differentially expressed in adipose tissue and 21 genes were differentially expressed in skeletal muscle. As per the criteria of Padj < 0.05 none of the T2DiACoD genes were significantly differentially expressed in patients’ liver tissues. 20 genes had FC values either >2 or <0.5. Of these, 20 genes, 1 is in adipose tissue and 19 are in skeletal muscle. We sought information on the sub-cellular location of the proteins encoded by the differentially expressed genes. Among those that were annotated as secreted, we further interrogated for their presence in plasma through text mining. In the following, only the differentially expressed genes at Padj < 0.05 are presented.
Cardiovascular
Among the genes implicated in cardiovascular complications, there are 56 genes differentially expressed in T2DM patients. These include 49 genes in pancreas, 8 genes in adipose tissue and 4 genes in skeletal muscle. Genes FABP5, SLC2A1 and TXN were differentially expressed in pancreas and adipose tissue whereas CD9 and TXN were differentially expressed in Pancreas and skeletal muscle. Seven differentially expressed genes CST3, FGF21, IL18, INS, and RBP4 are annotated as secreted and their product proteins are reportedly present in plasma134,135,136,137,138,139,140,141,142.
Atherosclerosis
Among the genes implicated in atherosclerotic complications, there are 37 genes differentially expressed in T2DM patients. These include 28 genes in pancreas, 8 genes in adipose tissue and 4 genes in skeletal muscle. Genes TXN and FABP5 were differentially expressed in adipose and pancreas. TXN was differentially expressed in all pancreas, adipose and skeletal muscle. Nine differentially expressed genes FGF21, IL18, INS, LEP, RBP4, ANGPTL2, MIF and VEGFA are annotated as secreted and are reportedly present in plasma136,137,138,139,140, 143,144,145. Two of these genes ANGPTL2 and VEGFA had FC values either >2 or <0.5.
Nephropathy
Among the genes implicated in nephropathy complications, 151 genes were differentially expressed in T2DM patients. These include 122 genes in pancreas, 26 genes in adipose tissue and 12 genes in skeletal muscle. The bar plots of the differentially expressed genes were generated to provide a graphical view of normalized gene expression in individual samples in terms of MAD modified Z-scores of diabetic and non-diabetic patients’ (Fig. 6). The y axis in the bar plots represents the normalized expression value whereas ‘x’ axis describes the samples. Additionally, differently expressed genes with P Value < 0.05 were also displayed liberally with an aim to benefit future studies. Genes ATF4, CCL5, EDNRA, SGK3, IGF2, SLC2A1, SOCS3, SRC and TIMM44 were differentially expressed in adipose and pancreas. 14 differentially expressed genes FGF21, IL18, RBP4, ANGPTL2, MIF, VEGFA, ANGPTL4, APOC1, CTGF, CXCL10, GAS6, GREM1, IFNG, IGF2, NGF, REG1A and TIMP2 136,137,138, 140, 141, 143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158 are annotated as secreted and their product proteins are reportedly present in plasma. Four of these genes VEGFA, TIMP2 and ANGPTL2 had FC values either >2 or <0.5.

Number of differentially expressed genes with padj < =0.05 in T2DM patient’s different tissues namely Pancreas, Skeletal muscle and Adipose tissue.
Neuropathy
Among the genes implicated in neuropathy complications, 39 genes were differentially expressed in T2DM patients’. These include 32 genes in pancreas, 7 genes in adipose tissue and 3 genes in skeletal muscle. IGF2 and TSPO were differentially expressed in adipose and pancreas. IRAK1 was differentially expressed in pancreas and skeletal muscle. Nine differentially expressed genes LEP, MIF, VEGFA, CXCL10, IFNG, IGF2, and NGF are annotated as secreted and their product proteins are also reportedly present in plasma139, 143, 144, 148, 150, 153,154,155,156. One of these genes, VEGFA had FC values either >2 or <0.5.
Retinopathy
Among the genes implicated in retinopathy complication 46 genes were differentially expressed in T2DM patients, 36 genes in pancreas, 9 genes in adipose tissue and 4 genes in skeletal muscle. Genes ATF4, IFG2 and SOCS3 were differentially expressed in adipose and pancreas. There are 10 differentially expressed genes FGF21, IL18, INS, RBP4, TIMP1, VEGFA, ANGPTL4, CTGF and IGF2 annotated as secreted and their product proteins are also reportedly present in plasma136,137,138,139,140, 145, 146, 149, 155. Two of these genes VEGFA and TIMP1 had FC values either >2 or <0.5.
Risk factors
We investigated the interaction of risk factors, namely, obesity, inflammation, diet and stress, with the genes of T2DM associated complications, which are differentially expressed in T2DM patients’ 3 tissues. The results are shown in Supplementary Figure 1.
In Pancreas, obesity topped the list with the number of differentially expressed genes (DEG) associated with the complications nephropathy and neuropathy, cardiovascular, and atherosclerosis whereas in case of retinopathy, inflammation topped the list (Supplementary Figure 1a).
In Adipose tissue, obesity topped the list with the number of DEG associated with nephropathy, atherosclerosis, whereas in neuropathy and retinopathy, stress and inflammation are at the top and in cardiovascular complication, diet and inflammation topped the list (Supplementary Figure 1b).
In Skeletal muscle, the number of DEG is less compared with Adipose and Pancreas. It is noteworthy that obesity topped the list with the number of DEG with nephropathy (Supplementary Figure 1c).
Mixed evidence on genes with passage of time
6 genes ACE, APOE, VEGFA, TNFRSF11B, FABP2 and VWF had mixed evidences and observations. In general, the associations of these genes were investigated multiple times. The results are presented in Supplementary Table 4b. It is evident that with time the associative evidences change however in some cases they also get reinforced. For example in the case of ACE’s role in cardiovascular, there is positive evidence in 1995 and 2003 but in 2005 in Chinese population a negative evidence was observed. On the other hand the role in Nephropathy, in 1995 there was negative evidence overturned in 2005 and later supported in as recent as 2015. Another noteworthy example is in case of VEGFA. The role of VEGFA in Retinopathy and Nephropathy was negative in several populations but reported positive in Caucasian (Retinopathy) in the years as recently as 2013–2015. Likewise in the case of TNFRS11B, negative evidences were documented in the years 2011 and 2013 but overturned in 2015 in its association with Atherosclerosis. These mixed observations appear most likely due to population specific effects and therefore our genes collection using a greedy approach offers resource for further investigation in a population specific manner. It is also noteworthy that the genes ACE, APOE, FABP2, TNFRS11B, VEGFA and VWF are targets of many commonly used drugs. The CLOCK gene was differentially expressed in both adipose tissue and pancreas in different studies.
Transcription factor binding sites and miRNA binding sites
We sought to decipher the RNA regulatory entities including transcription factor binding sites and miRNA target sites using RegRNA 2.0. Of 650 genes, 340 genes had 1351 sequence variants. Among the remaining genes, 306 genes had no variants. In case of 4 genes GGT2, NYS3, SAA@ and SERPI3 RefSeq DNA sequences were not available.
There were 1039 miRNA target sites encompassing 1272 variant sequences of 340 genes and 298 genes without variant sequences. There were 654 transcriptional regulatory motifs encompassing 1337 out of 1351 variant sequences of 340 genes and 268 genes without variant sequences. Of 7 genes (AGER, TNFRSF11B, CRK, PON1, ADIPOQ, CRP and NOS3) common to all complications, 3 genes (HMGB1, PON1 and TNFRSF11B) have no variant sequences and 4 genes (NOS3, CRK, AGER and ADIPOQ) have 17 variant sequences. 170 Transcriptional regulatory motifs and 6 miRNA target sites are present in all the 3 genes (HMGB1, PON1 and TNFRSF11B) without variant sequences. In the case of remaining 4 genes with 17 variant sequences there were 262 Transcriptional regulatory motifs and 43 miRNA target sites.
Among 9 genes (TNF, IGF2, TGFB1, IL1B, IL6, INS, GHR, APOE and EGF) with wide functional roles, 5 genes (IL6, INS, TGFB1, IL1B and TNF) had no variant sequences and 4 genes (APOE, EGF, GHR, and IGF2) had 25 variant sequences. The genes without variant sequences had 181 Transcriptional regulatory motifs and 19 miRNA target sites. Among 25 variant sequences of 4 genes there were 312 Transcriptional regulatory motifs and 38 miRNA target sites. In general, genes with variant had larger numbers of transcriptional regulatory motifs and miRNA target sites indicating larger regulatory space for these genes.
Discussion
T2DM, the most common form of diabetes principally arising due to insulin resistance and reduced insulin activity is a risk for several complications, namely atherosclerosis, neuropathy, nephropathy, retinopathy and cardiovascular diseases. The principal driver is the exposure of the cells, tissues and other proteins to hyperglycaemic conditions. Our goal in this work was to organize the dispersed information on the genes reportedly associated with complications in T2DM conditions accumulating in the literature. The T2DiACoD houses the compiled information and is made available to users to facilitate ongoing research in this area. Compared with other repositories developed so far towards similar ends, T2DiACoD is more comprehensive encompassing highest number of genes up to date, including information on T2DM complications and is devoid of false positive evidences because of inclusion of a final manual curation step. We believe a combined approach including automated text mining followed by final manual curation would serve better for data mining from large corpii.
The information from the literature have been collected from various sources, namely candidate gene studies, GWAS, miRNAs associated with T2DM and differential expression analysis of genes in T2DM patients’ tissues - pancreas, skeletal muscle, liver and adipose from publicly available data repositories.
The identification of genes studied either in a single or in multiple years bring out the significance of the gene in the particular disease. It is evident that in all complications the genes studied in multiple years outnumber those studied in a single year by 1.06–4.16 folds except in neuropathy, where the numbers in both categories are nearly equal. This analysis aids in recognition of the importance of the gene, namely, whether the gene is being studied for further information or no further analysis are required to illuminate the implication of this gene in the complication. Among the 23 drug targets, 13 have been studied in multiple years, two have been studied in a single year. In case of markers or biomarkers, 2 genes each were studied in multiple years as well as in a single year.
If a gene had high BWI several years ago but later not reported, then its significance may have been lost due to multiple reasons. Given that different genes emerge as significant in different populations such patterns are likely. Genes reported in multiple years usually are considered as reference mentioned in background or in comparison in results. It is worth noting the context of the genes reported in multiple years to guide future studies.
The GO analysis elaborates the distinctness of each complication associated with T2DM. Further it is apparent from the GO enrichment map that atherosclerosis and cardiovascular maps are similar and so are the maps of retinopathy and neuropathy. The nephropathy enrichment map appears distinct with chemokine activities. These results could inform future studies in regard to developing new therapeutic approaches by targeting the prominent pathways.
The genes AGER, TNFRSF11B, CRK, PON1, ADIPOQ, CRP and NOS3 are associated with all 5 major complications of T2DM but none of them attained fold change (FC) either >2 or <0.5 in any of the T2DM patients. It is probable that these genes perhaps play mediatory role as opposed to providing driving motive force. The added feature of T2DiACoD is to enlist the differentially expressed genes involved in the pathogenesis of complications in T2DM patients’ tissues thereby reporting up-regulated and down-regulated genes. These observations illuminate the progress of complications in T2DM patients’ tissues and could inform future studies designed to monitor such progression.
A particularly important data gained from this exercise is the involvement of multiple tissues with respect to differential gene expression of a few genes associated with T2DM complications. It is probable that the differential regulation in multiple tissues may underlie the debilitating complications of T2DM. When considered at high FC (either >2 or <0.5), the differentially expressed genes associated with T2DM complications was most observed in skeletal muscle. However, when all differentially expressed genes are considered that were statistically significant, pancreas tissue topped the list followed by adipose and skeletal muscle. These observations show that while the focus of T2DM complications are at the skeletal muscle tissues, a great number of changes in gene expression take place in pancreas and adipose tissues. The liver appears particularly aloof from these noticeable changes. Although the methods of observing gene expression differences are largely statistics based, it is evident from this work that multiple tissues are involved at apparently variable extents.
It is noteworthy that about a quarter of the protective genes (19 out of 63) are differentially expressed in T2DM patients’ tissues. These observations indicate that the influence on the differential expression of protective genes already starts in T2DM conditions. It is noteworthy that 29 of the genes associated with T2DM in various populations were observed as differentially expressed in T2DM patients’ tissues. These observations while highlighting the immense beneficial contribution of GWAS are also informative in that the gene expression differences between T2DM patients and healthy controls likely depend on additional as yet unknown factors that are likely variable. Among the differentially expressed genes VEGFA evidently appears striking and is associated with several complications of T2DM. This information emerging from the integrative approach used in this work could benefit further therapeutic approaches.
Another principal goal in our work was to mine the information of multiple tissues involvement and presence of the associated gene products in plasma. It is noteworthy that a minority of associated genes were differentially expressed in multiple tissues and a minority of genes were reported as detected in plasma. These observations and value added information could serve for marker development in future well designed studies. The differential expressed genes and their product proteins of TIMP1, ANGPTL2, VEGFA, and TIMP2, reportedly present in plasma could be measured in different stages of T2DM complications and possibly offer information for clinical interventions.
The RegRNA 2.0 data analysis predicted that nearly half of the T2DM genes associated with complications had a large number of variants with proportionately large number of transcription factor binding sites and miRNA binding sites. Although only 34 T2DM associated miRNA targets overlapped with the genes in T2DiACoD database, it is quite probable that a large number of miRNAs may be involved in gene regulation system. This appears to increase the complexity of the entire system of genes underlying the T2DM complications. These data serve to highlight the complex nature of these disorders. However, it is noteworthy that a handful of miRNAs and genes have been identified for therapeutic effects.
Among the risk factors considered in this work, are obesity, inflammation, stress and diet and their interactions with the genes of T2DM associated complications. The major share of differentially expressed genes in T2DM patients as observed here is contributed by pancreas tissue followed by adipose and skeletal muscle tissues. Obesity is clearly a dominant risk factor interacting with the genes of T2DM complications followed by inflammation, diet and stress to variable extents. These results indicate the likelihood of developing a complication given the tissue of measurement of differentially expressed genes and the contributing risk factor. However, it is to be noted that the interaction of the risk factor with a given gene and the association of the same gene with T2DM complication were usually observed from different reports. Thus, these indications provide leads for designing specific future studies.
Thus it is apparent that obesity and inflammation play major role in the development of T2DM and its associated complications and therefore therapeutic regimens may be targeted towards ameliorating these effects. The control of obesity is recommended in the MedlinePlus159 site also.
Conclusions
Several points have emerged from our integrative analysis. (1) We believe a combined approach including automated text mining followed by final manual curation would serve better for data mining from large corpii. (2) It is evident that in all complications the genes studied in multiple years outnumber those studied in a single year by 1.06–4.16 folds except in neuropathy, where the numbers in both categories are nearly equal. This trends analysis shows that many genes are studied by different groups in multiple years meaning that the genes are reliably treated. (3) Among the 23 drug targets, 13 have been studied in multiple years, two have been studied in a single year. In case of markers or biomarkers 2 genes each were studied in multiple years as well as in a single year. Thus majority of the drug targets receive importance in investigations. (4) Genes reported in multiple years usually are considered as reference mentioned in background or in comparison in results. It is worth noting the context of the genes reported in multiple years to guide future studies. (5) It is apparent from the GO enrichment map that atherosclerosis and cardiovascular maps are similar and so are the maps of retinopathy and neuropathy. The nephropathy enrichment map appears distinct with chemokine activities. (6) A few genes ACE2, ADCYAP1, HDAC4, NCF1, NFE2L2, OSM, SMAD1, TGFB1, BDNF, SYVN1, TXNIP, CD36, CYP2J2, NLRP3, where the details of protective role are known have been described are displayed on the website. (7) Among the differentially expressed genes, VEGFA evidently appears striking and is associated with several complications of T2DM. This information emerging from the integrative approach used in this work could benefit further therapeutic approaches. (8) The identification of differentially expressed genes associated with T2DM and its complications in T2DM patients compared with normals indicate that the conditions for setting in of these complications arise in T2DM state already. (9) These observations indicate that the influence on the differential expression of protective genes already starts in T2DM conditions. (10) Obesity is clearly a dominant risk factor interacting with the genes of T2DM complications followed by inflammation, diet and stress to variable extents.
Material and Methods
All text analytics were carried out in R 3.0.1 and the CRAN package pubmed.mineR160. Additional packages161 were also used to supplement. An R package wordcloud was used for viewing terms in their order of occurrence frequencies.
Text mining and curation
Classification, summarization and additional data extraction
A total of 4,46,438 abstracts were extracted using the keyword “diabetes” from the PubMed database till 30th November 2016. This corpus is referred to as primary corpus. Wordcloud analysis using the R package “wordcloud“162 of the top ranking words extracted using the R package pubmed.mineR revealed that among the complications in diabetic conditions, the top ranking terms were atherosclerosis, neuropathy, nephropathy, retinopathy and cardiovascular and therefore we sub-classified the primary corpus into these 5 sub-corpora using the package pubmed.mineR160.
Population Study
Among the ethnic population terms, the frequently occurring terms were Indian (excluding Indian Americans), Japanese, Chinese, Americans (African Americans, Indian Americans, White Americans and Asian Americans), European, and Mexican. We used these terms for sub-classification of the primary corpus. The complete information about the gene including SNP information in different populations was obtained using the package NCBI2R163. In addition, we collected SNP IDs (rsIDs), their genomic position, odds ratio (OR), and reported p-values in GWAS.
Entity recognition and their relationships
Towards a comprehensive approach for gene data mining from literature texts we used sentence tokenization function of pubmed.mineR to extract sentences with co-occurrence of the entities, namely, gene symbols including official symbols recommended by the Human Gene Nomenclature Committee’s (HGNC), other symbols (aliases, previous symbols), alternative names from UniProt and gene name from HGNC and the disease complication terms and their aliases, using sentence tokenization of pubmed.mineR. Because the search algorithm uses regular expression based matching, false positives arise due to use of identical acronyms by authors signifying other meanings. Therefore the extracted sentences were examined for ‘proof of association’ or ‘of evidence’ by examining the entities and the relationship between them. The exercise was repeated with risk factors, namely, obesity, stress (oxidative stress and endoplasmic reticulum stress), diet and inflammation in order to identify lateral associations. These risk factors were top ranking based on their occurrence frequencies. In cases where the assertion was not clear, the entire abstract was examined along with full text wherever available from PubMed central. We used a liberal approach in collecting both ‘firm’ assertions and ‘likely’ indications. [Supplementary: source code].
Trends analysis
The Buzz Word Index (BWI) proposed by Jensen et al.164 provides a convenient approach to obtain the trends in the literature. We computed the BWI value of genes using pubmed.mineR. Buzz words are terms mentioned frequently in a given year compared to that in previous years. In general, if an abstract in a given year contains repeated mention of a gene due to its detailed characterization carried out in the study and reported compared to all the abstracts texts in previous years, the BWI registers a high value. We used a minimum of BWI = 1 in order to collect all the genes attributed with some importance. A single occurrence of a given gene in a given year would have its corresponding BWI register “zero” in that year.
Data Collection from other databases
We compared our T2DiACoD with other available databases for T2D and associated complications, namely, DisGeNET and T2D-Db (Table 2) and collected the genes missed in our data mining approach. We obtained two genes for diabetic nephropathy from T2D-Db whereas from DisGeNET we got 16 genes for diabetic nephropathy and 10 genes for diabetic retinopathy. Other genes in these databases were false positives verified by examining the evidences and references they reported and therefore they were excluded.
Web server development
The web interface of T2DiACoD was developed using PHP 5.6.3 and HTML. The consolidated data were entered in MySQL 5.0.11 tables using XAMPP (v3.1.0). XAMPP is an open source cross platform package. It includes MariaDB (database), Apache 2.4.10 (server application) and PHP (scripting language). Data sets can be downloaded as Excel sheets.
Database structure and content
T2DiACoD database structure consists of diabetic complications, ethnicity (population) studies including GWAS in T2DM, and Drug targets of recent drug development from the literature. Differentially Expressed genes in patients’ and normals’ tissues including adipose, pancreas, and skeletal muscle and miRNAs with target in T2DM and its complications are included. The risk factors included are obesity, inflammation, diet and stress (oxidative stress & endoplasmic reticulum stress). In the case of complications in T2DM cases, we considered atherosclerosis, cardiovascular disease, nephropathy, neuropathy and retinopathy. The information on genes obtained from the respective sub-classified corpora were cast into three tables: (i) Gene information: gene symbol, gene name, synonyms, chromosome number, homologene id in a given complication (ii) NCBI Gene database, (iii) Summary of individual genes: Locus id, gene symbol, gene name, synonyms, Chromosome, Homologene id and observation reporting evidence, and the corresponding PubMed ID (PMID) of the report. The population block contains genes associated with diabetes in various populations and sub-classified as reported and includes SNP ids (rsIds), risk allele, chromosome number, chromosome position, functional class, p-value, odds-ratio and PMID.
Expression tab
It consists of graphical view (histogram display) of the differential gene expression in adipose, pancreatic, and skeletal muscle tissues in human patients’. The raw (.CEL file format for affymetrix data or BeadStudio output file format for illumina data) microarray gene expression data for different tissues were accessed from the NCBI GEO165. We used different combinations of keywords such as ‘type 2 diabetes AND pancreas’, ’type 2 diabetes AND liver’, ‘type 2 diabetes AND skeletal muscle’, ‘type 2 diabetes AND adipose’ for data retrieval. We collected a total of 11 datasets (GSE29226, GSE40234, GSE16415, GSE23343, GSE15653, GSE25724, GSE38642, GSE20966, GSE29221, GSE25462, GSE12643) for all the available tissues studied in different populations. The affymetrix data were pre-processed using the affy package166. The .CEL files for each GSE id was read using ReadAffy() and subsequently we performed the background correction and computed the expression using the robust mean average (rma()) function with ‘normalize = FALSE’, which computes the expression and performs the background correction. The Illumina data were pre-processed using lumi package167 of Bioconductor. The lumi package was used to read the illumina BeadStudio output file for each GSE id using the lumiR() function followed by background correction and computed the expression using the robust mean average (rma()) function with ‘normalize = FALSE’, which computes the expression and performs the background correction. Subsequently, the expression of probes with detection p-values < 0.05 was retained to eliminate low signal intensity genes. The pre-processed data was transformed individually into standard normal using the modified Z-score method based on median absolute deviation (MAD)168. MAD measures the variability in the sample about its median. Each dataset was pre-processed and normalised individually (Supplementary Table 5).
where, Xi = data element of the ith row
Xj = median of the jth column
MADj = Median Absolute deviation of jth column
Mi = Modified Z-score
Expression of duplicated genes were averaged using the CollapseRows() function of whole genome co-expression network analysis (WGCNA) R package169. The rows containing excessive amount of missing data (90% or more) were omitted. The differentially expressed genes of the normalized data were computed between non-diabetic and diabetic human patients’ tissues (Supplementary Figure 2[a-i]). The limma package161 was used to compute differential expression. Empirical Bayes moderated t-statistics test was applied to test each individual contrast equal to zero. The topTable() function was used to rank genes in order of evidence for differential expression. The genes with P-adjusted (Benjamini and Hochberg) values < 0.05 were selected as differentially expressed genes and their P Values were also noted.
miRNA block
miRNAs are small non-coding RNA molecules of 20–25 nt that negatively regulate translation of the target mRNAs by binding to the 3′UTR region170. Several miRNAs have been identified that are likely involved in T2DM and their complications170. The miRNAs associated with T2DM were identified from the literature along with their corresponding target genes. Subsequently, the targets of these miRNAs were mapped with the gene list of T2DM complications to identify miRNAs with potential for regulation of genes associated with T2DM complications.
Functional enrichment analysis
To infer potential biological significance of the reported genes we have used DAVID (Database for Annotation, Visualization and Integrated Discovery)171. It provides functional annotation and analysis of gene list. The lists of genes were submitted to functional annotation in DAVID to analyze the Gene Ontology (GO), protein domains and pathways. The relevant GO terms associated with input gene list were extracted and analyzed. The GO structure mainly contains three categories, namely, Biological Process (BP) for biological goals, Molecular Function (MF) describing functional roles and Cellular Component (CC) indicates the cellular location where gene products are active. The output table was obtained with the following information: Annotation Cluster, Enrichment Score, Category, Term, Count, % (involved genes/total genes), PValue, Genes, List Total, Pop Hits, Pop Total, Fold Enrichment, Bonferroni, Benjamini and FDR. P-Value (calculated by Fishers’ exact test) as well as FDR< = 0.05 were considered strongly enriched in the annotation categories. The cytoscape plugin EnrichmentMap was used for graphical display of GO terms172. EnrichmentMap is a visualisation method for the gene set enrichment results. In the graph, nodes represent gene set involved in the GO term and edges represent the extent of overlap of genes between the two gene sets connected by them. Node size and edge width are proportional to the number of genes. The connection between two nodes describes set of genes that two nodes have in common.
Enriched or dilution of positive genes
We selected a sample of 96 genes (Supplementary Table 4a), which were described to be strongly associated with any of the 5 given complications in T2DM through manual examination. We examined whether published reports emerged refuting their associative assertions. Official HGNC symbols as well as alias, previous symbol and full names were considered. In the next steps, the sentences with the word “not” were extracted. The pattern match in the regular expression function was used with the pattern “not” prefixed and suffixed with a space to ensure high specificity. A total of 6 genes with sentences having the word “not” in their observation were identified along with their PMIDs.
As an example: The Ala54Thr polymorphism of the FABP2 gene is not associated with CHD, markers of the metabolic syndrome, or the fatty acid profile of serum lipids in Finnish CHD patients.PMID:12189904. For the same gene the positive evidence in our database is FABP2 confers susceptibility to renal disease in type 2 diabetic patients.PMID:16249461.
RegRNA Analysis
Functional RNA motifs can be identified in input mRNA sequences using the RegRNA 2.0 integrated web server173,174,175,176,177,178,179,180. It provides information on Splice sites (donor site, acceptor site, Splicing regulatory motifs (ESE, ESS, ISE, ISS elements)173, Polyadenylation sites, Transcriptional motifs (rho-independent terminator, TRANSFAC)174, Translational motifs (ribosome binding sites), UTR motifs (UTRsite patterns)175, mRNA degradation elements (AU-rich elements), RNA editing sites (C-to-U editing sites), Riboswitches (RiboSW)176, RNA cis-regulatory elements (Rfam, ERPIN)177, similar functional RNA sequences (fRNAdb)178, RNA-RNA interaction regions (miRNA, ncRNA)178, User defined Motif (RNAMotif)179, Miscellaneous information (open reading frame, GC-ratio, RNA accessibility)180 Sequences greater than 12 kbp size were divided into two equal halves and RegRNA analysis carried out and considered in overall counts.
Sub-cellular location
Subcellular locations for all the genes in T2DIACoD were collected from the UniProt181.
Data access for users
Users can access T2DiACoD by mouse click on the complications in diabetic conditions (atherosclerosis, diabetic neuropathy, diabetic nephropathy, diabetic retinopathy, cardiovascular disease) and Drug targets. The gene centric information for all the genes enlisted for the corresponding ‘selection’ will be displayed. Additional information on gene ontology, literature reference reporting evidence is also provided. The population studies (Chinese, American, Indian, European, Japanese, and Mexican) link enables the user to access the single nucleotide polymorphism (SNP) information on the variations investigated in different ethnic populations with T2DM including link to the published references reporting the data.
The expression search tab on the homepage offers graphical display of the differentially expressed genes in different patients’ tissues – adipose, pancreas, and skeletal muscle.
We have provided the expression tables of each gene for four different tissues (Adipose, Skeletal Muscle, and Pancreas) having different sample Ids with their corresponding-value,adj.P-value and fold change (FC) values as well as brief information of microarray data and its sample. The user can also view the expression graph by clicking the expression link from the gene and SNP information data tables. The bar graph displays the gene expression in diabetic cases compared to the non-diabetic controls in different tissues. A liberal selection was adopted including genes differentially expressed at Padj < = 0.05 and p value < = 0.05 for visual inspection by users and to guide future studies. The miRNA search tab provides information about the miRNAs with their corresponding target gene associated with T2DM and the link to the reference describing evidential support of miRNAs to the pathogenesis of disease.
Instant search option with gene symbol is available to obtain a brief summary of the genes including Locus id, gene symbol, gene name, synonyms, Chromosome, Homologene id and observation reporting evidence with corresponding literature reference PubMed ID (PMID). An Advanced search is also made available to conduct combinatorial queries to fetch the common genes between T2DM associated complex disorders and risk factors, namely, stress, inflammation, diet and obesity known to predispose individuals to T2DM.
References
Ashcroft, F. M. & Rorsman, P. Diabetes mellitus and the beta cell: The last ten years. Cell 148, 1160–1171 (2012).
Prasad, R. B. & Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel). 6, 87–123 (2015).
Patel, C. J. et al. Systematic identification of interaction effects between genome-and environment-wide associations in type 2 diabetes mellitus. Human genetics 132(5), 495–508 (2013).
Schwartz, D. & Collins, F. Environmental biology and human disease. Science 316(5825), 695–696 (2007).
Einstein: Albert Einstein College of Medicine - diabetes facts and statistics. https://www.einstein.yu.edu/centers/diabetes-research/facts-statistics/ Date of Access: 20/11/2015 (2015).
Miloš, S. et al. Repeated low-dose 17β-estradiol treatment prevents activation of apoptotic signaling both in the synaptosomal and cellular fraction in rat prefrontal cortex following cerebral ischemia. Neurochemistry international 83, 1–8 (2015).
Prokopenko, I., McCarthy, M. I. & Lindgren, C. M. Type 2 diabetes: new genes, new understanding. Trends Genet. 24, 613–21 (2008).
Teslovich, T. M. et al. Re-sequencing expands our understanding of the phenotypic impact of variants at GWAS loci. PLoS Genet 10, 1 (2014).
Patel, C. J., Chen, R., Kodama, K., Ioannidis, J. P. A. & Butte, A. J. Systematic identification of interaction effects between validated genome- and environment-wide associations on Type 2 Diabetes Mellitus. AMIA Jt. Summits Transl. Sci. Proc. AMIA Summit Transl. Sci. 2013, 135 (2013).
Lyssenko, V. et al. Genetic screening for the risk of type 2 diabetes: worthless or valuable? Diabetes Care 36(Suppl 2), S120–6 (2013).
Palmer, N. D. et al. A genome-wide association search for type 2 diabetes genes in african americans. PLoS One 7 (2012).
Wheeler, E. & Barroso, I. Genome-wide association studies and type 2 diabetes. Brief. Funct. Genomics 10, 52–60 (2011).
Hakonarson, H. & Grant, S. F. Genome‐wide association studies (GWAS): impact on elucidating the aetiology of diabetes. Diabetes/metabolism research and reviews 27(7), 685–696 (2011).
Fuchsberger, C. et al. The genetic architecture of type 2 diabetes. Nature 536, 41–7 (2016).
Sreekumar, R., Halvatsiotis, P., Schimke, J. C. & Sreekumaran Nair, K. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 51, 1913–1920 (2002).
Nair, S. et al. Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia 48(9), 1784–1788 (2005).
Zelezniak, A., Pers, T. H., Soares, S., Patti, M. E. & Patil, K. R. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput. Biol. 6, e1000729 (2010).
Sengupta, U., Ukil, S., Dimitrova, N. & Agrawal, S. Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PloS one 4(12), e8100 (2009).
Zhong, H. et al. Liver and Adipose Expression Associated SNPs Are Enriched for Association to Type 2 Diabetes. Plos Genet. 6, 12 (2010).
Herzig, S. Liver: A target of late diabetic complications. Experimental and Clinical Endocrinology and Diabetes 120, 202–204 (2012).
Shore, A. C. et al. Measures of atherosclerotic burden are associated with clinically manifest cardiovascular disease in type 2 diabetes: A European cross-sectional study. J. Intern. Med. 278, 291–302 (2015).
Korish, A. A. et al. Camel milk attenuates the biochemical and morphological features of diabetic nephropathy: inhibition of Smad1 and collagen type IV synthesis. Chemico-biological interactions 229, 100–108 (2015).
Gandhi, C. R. & Ross, D. H. Microwave induced stimulation of 32Pi incorporation into phosphoinositides of rat brain synaptosomes. Radiat. Environ. Biophys. 28, 223–234 (1989).
Park, E. I. et al. Lipid level and type alter stearoyl CoA desaturase mRNA abundance differently in mice with distinct susceptibilities to diet-influenced diseases. J. Nutr. 127, 566–573 (1997).
Hegele, R. A. et al. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. The Lancet Diabetes and Endocrinology 2, 655–666 (2014).
Carpena, M. P. et al. Genetics of diabetic nephropathy. Arquivos Brasileiros de Endocrinologia & Metabologia 54(3), 253–261 (2010).
Gohda, T. et al. Genetic susceptibility to type 2 diabetic nephropathy in human and animal models. Nephrology 10(s2), S22–S25 (2005).
Dobrowsky, R. T., Rouen, S. & Yu, C. Altered neurotrophism in diabetic neuropathy: spelunking the caves of peripheral nerve. J. Pharmacol. Exp. Ther. 313, 485–491 (2005).
Arar, N. H. et al. Heritability of the severity of diabetic retinopathy: The FIND-Eye study. Investig. Ophthalmol. Vis. Sci. 49, 3839–3845 (2008).
Narne, P., Ponnaluri, K. C., Siraj, M. & Ishaq, M. Association Analysis of Polymorphisms in Genes Related to Oxidative Stress in South Indian Type 2 Diabetic Patients with Retinopathy. Ophthalmic Genet. 1–8, doi:10.3109/13816810.2014.895015 (2014).
Bessueille, L. & Magne, D. Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell. Mol. Life Sci. 72, 2475–2489 (2015).
Yoo, H. J. & Choi, K. M. Hepatokines as a link between obesity and cardiovascular diseases. Diabetes and Metabolism Journal 39, 10–15 (2015).
Agrawal, S. et al. T2D-Db: an integrated platform to study the molecular basis of Type 2 diabetes. BMC genomics 9(1), 320 (2008).
Lim, J. E. et al. Type 2 diabetes genetic association database manually curated for the study design and odds ratio. BMC medical informatics and decision making 10(1), 76 (2010).
Dai, H. J., Wu, J. C. Y., Tsai, R. T. H., Pan, W. H. & Hsu, W. L. T-HOD: A literature-based candidate gene database for hypertension, obesity and diabetes. Database 2013 (2013).
Piñero, J. et al. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. http://www.disgenet.org/web/DisGeNET/menu Database, Date of access: 13/01/2016 (2015).
Agrawal, S. et al. T2D-Db: an integrated platform to study the molecular basis of Type 2 diabetes. http://t2ddb.ibab.ac.in/home.shtml BMC genomics 9(1), 320, Date of Access: 13/01/2016 (2008).
T2D-GENES Consortium, GoT2D Consortium, DIAGRAM Consortium. 2017, May, Available at http://www.type2diabetesgenetics.org/.
Li, W. B. et al. Silencing of activin receptor-like kinase 7 alleviates aortic stiffness in type 2 diabetic rats. Acta Diabetol. 52, 717–726 (2015).
Connelly, P. W. et al. Association of the novel cardiovascular risk factors paraoxonase 1 and cystatin C in type 2 diabetes. J. Lipid Res. 50, 1216–1222 (2009).
Barathi, S., Charanya, M., Muthukumaran, S., Angayarkanni, N. & Umashankar, V. Comparative modeling of PON2 and analysis of its substrate binding interactions using computational methods. J. Ocul. Biol. Dis. Infor. 3, 64–72 (2010).
Ikeda, Y. et al. Low human paraoxonase predicts cardiovascular events in Japanese patients with type 2 diabetes. Acta Diabetol. 46, 239–242 (2009).
Odeberg, J., Larsson, C. A., Rastam, L. & Lindblad, U. The Asp(298) allele of endothelial nitric oxide synthase is a risk factor for myocardial infarction among patients with type 2 diabetes mellitus. BMC Cardiovasc. Disord. 8 (2008).
Blázquez‐Medela, A. M. et al. Osteoprotegerin is associated with cardiovascular risk in hypertension and/or diabetes. European journal of clinical investigation 42(5), 548–556 (2012).
Katakami, N. et al. Adiponectin G276T gene polymorphism is associated with cardiovascular disease in Japanese patients with type 2 diabetes. Atherosclerosis 220(2), 437–442 (2012).
Yan, X. X. et al. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 205(2), 544–548 (2009).
Strawbridge, R. J. et al. serum 25-hydroxyvitamin D concentration-associated genetic variant in DHCR7 interacts with type 2 diabetes status to influence subclinical atherosclerosis (measured by carotid intima–media thickness). Diabetologia 57(6), 1159–1172 (2014).
Kim, M. K. et al. Clinical utility of serum beta-2-microglobulin as a predictor of diabetic complications in patients with type 2 diabetes without renal impairment. Diabetes & metabolism 40(6), 459–465 (2014).
Chang, Y. C. et al. Hyperglycemia accelerates ATP‐binding cassette transporter A1 degradation via an ERK‐dependent pathway in macrophages. Journal of cellular biochemistry 114(6), 1364–1373 (2013).
Oram, J. F. & Heinecke, J. W. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 85, 1343–1372 (2005).
Kyriakou, T. et al. Genotypic effect of the −565C >T polymorphism in the ABCA1 gene promoter on ABCA1 expression and severity of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 25(2), 418–423 (2005).
Sartippour, M. R. & Renier, G. Differential regulation of macrophage peroxisome proliferator-activated receptor expression by glucose: role of peroxisome proliferator-activated receptors in lipoprotein lipase gene expression. Arter. Thromb Vasc Biol 20, 104–110 (2000).
Wang, X. Q., Yang, K., He, Y. S., Lu, L. & Shen, W. F. Receptor mediated elevation in FABP4 levels by advanced glycation end products induces cholesterol and triacylglycerol accumulation in THP-1 macrophages. Lipids 46, 479–486 (2011).
Nakamura, A. et al. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes care 28(12), 2890–2895 (2005).
Zhang, W. et al. The functional Q84R polymorphism of TRIB3 gene is associated with diabetic nephropathy in Chinese type 2 diabetic patients. Gene 555(2), 357–361 (2015).
Zhang, L. et al. Down-regulation of miR-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. Journal of Diabetes and its Complications 28(3), 259–264 (2014).
Stefanidis, I. et al. Association Between the Interleukin-1β Gene (IL1B) C− 511T Polymorphism and the Risk of Diabetic Nephropathy in Type 2 Diabetes: A Candidate–Gene Association Study. DNA and cell biology 33(7), 463–468 (2014).
Hookham, M. B. et al. Insulin receptor substrate‐2 is expressed in kidney epithelium and up‐regulated in diabetic nephropathy. FEBS journal 280(14), 3232–3243 (2013).
Bonomo, J. A. et al. The ras responsive transcription factor RREB1 is a novel candidate gene for type 2 diabetes associated end-stage kidney disease. Human molecular genetics 23(24), 6441–6447 (2014).
Shcherbak, N. S., Shutskaya, Z. V., Sheidina, A. M., Larionova, V. I. & Schwartz, E. I. Methylenetetrahydrofolate reductase gene polymorphism as a risk factor for diabetic nephropathy in IDDM patients. Mol. Genet. Metab. 68, 375–378 (1999).
Mazza, A. et al. Serum homocysteine, MTHFR gene polymorphism, and carotid intimal-medial thickness in NIDDM subjects. Journal of thrombosis and thrombolysis 8(3), 207–212 (1999).
Abu El-Asrar, A. M., Mairaj Siddiquei, M., Nawaz, M. I., Geboes, K. & Mohammad, G. The proinflammatory cytokine high-mobility group box-1 mediates retinal neuropathy induced by diabetes. Mediators Inflamm. 2014 (2014).
Simon, C. M. et al. Dysregulated IGFBP5 expression causes axon degeneration and motoneuron loss in diabetic neuropathy. Acta neuropathologica 130(3), 373–387 (2015).
Hellweg, R. & Hartung, H. D. Endogenous levels of nerve growth factor (NGF) are altered in experimental diabetes mellitus: a possible role for NGF in the pathogenesis of diabetic neuropathy. Journal of neuroscience research 26(2), 258–267 (1990).
Faradji, V. & Sotelo, J. Low serum levels of nerve growth factor in diabetic neuropathy. Acta Neurol. Scand. 81, 402–406 (1990).
Hernández-Pedro, N. et al. Vitamin A increases nerve growth factor and retinoic acid receptor beta and improves diabetic neuropathy in rats. Translational Research 164(3), 196–201 (2014).
Burdon, K. P. et al. Genome-wide association study for sight-threatening diabetic retinopathy reveals association with genetic variation near the GRB2 gene. Diabetologia 58(10), 2288–2297 (2015).
Yamaji, Y. et al. TEM7 (PLXDC1) in neovascular endothelial cells of fibrovascular membranes from patients with proliferative diabetic retinopathy. Investigative ophthalmology & visual science 49(7), 3151–3157 (2008).
Pradeepa, R. et al. Association of serum adiponectin with diabetic microvascular complications among south Indian type 2 diabetic subjects - (CURES-133). Clin. Biochem. 48, 33–38 (2015).
Niu, W. et al. A meta-analysis of receptor for advanced glycation end products gene: Four well-evaluated polymorphisms with diabetes mellitus. Mol. Cell. Endocrinol. 358, 9–17 (2012).
Katakami, N. et al. Adiponectin G276T gene polymorphism is associated with cardiovascular disease in Japanese patients with type 2 diabetes. Atherosclerosis 220, 437–42 (2012).
Fredriksson, J. et al. Variation in GYS1 interacts with exercise and gender to predict cardiovascular mortality. PLoS One 2 (2007).
M., G. et al. The relationship of ACE and CETP gene polymorphisms with cardiovascular disease in a cohort of Asian Indian patients with and those without type 2 diabetes. J. Diabetes Complications 25, 303–308 (2011).
Wu, S. S. et al. The relationship of angiotensin I-converting enzyme gene polymorphism with diabetic retinopathy and diabetes myocardial infarction. Zhonghua yi xue yi chuan xue za zhi=Zhonghua yixue yichuanxue zazhi=Chinese journal of medical genetics 21(3), 283–285 (2004).
Ait-Oufella, H. et al. Recent advances on the role of cytokines in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 31(5), 969–979 (2011).
Solini, A., Santini, E., Nannipieri, M. & Ferrannini, E. High glucose and homocysteine synergistically affect the metalloproteinases-tissue inhibitors of metalloproteinases pattern, but not TGFB expression, in human fibroblasts. Diabetologia 49, 2499–2506 (2006).
Hodgkinson, C. P., Laxton, R. C., Patel, K. & Ye, S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 2275–2281 (2008).
Cooper, M. E. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet (London, England) 352, 213–9 (1998).
Cooper, M. E. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 44, 1957–1972 (2001).
Pálsson, R. & Patel, U. D. Cardiovascular complications of diabetic kidney disease. Adv. Chronic Kidney Dis. 21 (2014).
Ni, W. J., Tang, L. Q. & Wei, W. Research progress in signalling pathway in diabetic nephropathy. Diabetes/metabolism research and reviews 31(3), 221–233 (2015).
Raptis, A. E. & Viberti, G. Pathogenesis of diabetic nephropathy. Experimental and clinical endocrinology & diabetes: official journal, German Society of Endocrinology and German Diabetes Association 109, S424–37 (2000).
Luo, J. et al. TCF7L2 variation and proliferative diabetic retinopathy. Diabetes 62, 2613–2617 (2013).
Hughes, J. M. et al. Advanced glycation end products cause increased CCN family and extracellular matrix gene expression in the diabetic rodent retina. Diabetologia 50, 1089–1098 (2007).
Kaidonis, G. et al. Common Sequence Variation in the VEGFC Gene Is Associated with Diabetic Retinopathy and Diabetic Macular Edema. Ophthalmology 122, 1828–1836 (2015).
Østergaard, L. et al. The effects of capillary dysfunction on oxygen and glucose extraction in diabetic neuropathy. Diabetologia 58(4), 666–677 (2015).
Callaghan, B. C., Cheng, H. T., Stables, C. L., Smith, A. L. & Feldman, E. L. Diabetic neuropathy: Clinical manifestations and current treatments. The Lancet Neurology 11, 521–534 (2012).
Cameron, N. E., Eaton, S. E., Cotter, M. A. & Tesfaye, S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 44, 1973–1988 (2001).
Tsuzuki, S. et al. The examination of apoE phenotypes in diabetic patients with peripheral neuropathy. Rinsho Byori. 46, 829–833 (1998).
Vincent, A. M. et al. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp. Neurol. 208, 216–227 (2007).
Wang, L. et al. Phosphodiesterase-5 is a therapeutic target for peripheral neuropathy in diabetic mice. Neuroscience 193, 399–410 (2011).
Bhatia, H., Verma, G. & Datta, M. MiR-107 orchestrates ER stress induction and lipid accumulation by post-transcriptional regulation of fatty acid synthase in hepatocytes. Biochim. Biophys. Acta - Gene Regul. Mech. 1839, 334–343 (2014).
Wijesekara, N. et al. miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 61, 653–658 (2012).
Párrizas, M. et al. Circulating miR-192 and miR-193b are markers of prediabetes and are modulated by an exercise intervention. J. Clin. Endocrinol. Metab. 100, E407–E415 (2015).
Bang-Berthelsen, C. H. et al. Independent component and pathway-based analysis of miRNA-regulated gene expression in a model of type 1 diabetes. BMC Genomics 12, 97 (2011).
Kornfeld, J.-W. et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature 494, 111–5 (2013).
Higuchi, C. et al. Identification of circulating miR-101, miR-375 and miR-802 as biomarkers for type 2 diabetes. Metabolism 64(4), 489–497 (2015).
Olivieri, F. et al. MiR-21-5p and miR-126a-3p levels in plasma and circulating angiogenic cells: relationship with type 2 diabetes complications. Oncotarget 6, 35372–35382 (2015).
Ling, H. Y. et al. MiRNA-21 reverses high glucose and high insulin induced insulin resistance in 3T3-L1 adipocytes through targeting phosphatase and tensin homologue. Exp. Clin. Endocrinol. Diabetes 120, 553–559 (2012).
Zhou, B. et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 55, 2032–2043 (2012).
Locke, J. M., Lango Allen, H. & Harries, L. W. A rare SNP in pre-miR-34a is associated with increased levels of miR-34a in pancreatic beta cells. Acta Diabetol. 51, 325–329 (2014).
Corral-Fernández, N. E. et al. Dysregulated miR-155 expression in peripheral blood mononuclear cells from patients with type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 121, 347–53 (2013).
Miller, A. M. et al. MiR-155 Has a Protective Role in the Development of Non-Alcoholic Hepatosteatosis in Mice. PLoS One 8 (2013).
Sebastiani, G. et al. MicroRNA-124a is hyperexpressed in type 2 diabetic human pancreatic islets and negatively regulates insulin secretion. Acta Diabetol. 52, 523–530 (2015).
Karolina, D. S. et al. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS One 6 (2011).
Wang, X. et al. Determination of 14 circulating microRNAs in Swedes and Iraqis with and without diabetes mellitus type 2. PloS one 9(1) (2014).
Zhang, Y. et al. MicroRNA-106b induces mitochondrial dysfunction and insulin resistance in C2C12 myotubes by targeting mitofusin-2. Molecular and cellular endocrinology 381(1), 230–240 (2013).
Wu, Y. et al. Identification of miR-106b-93 as a negative regulator of brown adipocyte differentiation. Biochem. Biophys. Res. Commun. 438, 575–580 (2013).
Setyowati Karolina, D., Sepramaniam, S., Tan, H. Z., Armugam, A. & Jeyaseelan, K. miR-25 and miR-92a regulate insulin I biosynthesis in rats. RNA Biol. 10, 1365–78 (2013).
Hou, X., Wu, W., Yin, B., Liu, X. & Ren, F. MicroRNA‐463‐3p/ABCG4: A new axis in glucose‐stimulated insulin secretion. Obesity 24(11), 2368–2376 (2016).
Latouche, C. et al. MicroRNA-194 modulates glucose metabolism and its skeletal muscle expression is reduced in diabetes. PLoS One 11 (2016).
Yang, W. M., Jeong, H. J., Park, S. W. & Lee, W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res. 59, 2303–2314 (2015).
Shan, Q. et al. Epigenetic modification of miR-10a regulates renal damage by targeting CREB1 in type 2 diabetes mellitus. Toxicol. Appl. Pharmacol. 306, 134–143 (2016).
Luo, B. et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One 9(8) (2014).
Ma, B. et al. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology 154(8), 2843–2856 (2013).
Geloen, A. et al. CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS One 7(5), e37633 (2012).
Zheng, H. et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 60(11), 3055–3066 (2011).
Fan, J. & Xu, G. Heme oxygenase-1 mediates Nrf2-dependent protection of neurons and vascular endothelial cells in diabetic retinopathy. Investigative Ophthalmology & Visual Science 52(14), 4446–4446 (2011).
Negi, G., Kumar, A. & Sharma, S. S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF‐κB and Nrf2 cascades. Journal of pineal research 50(2), 124–131 (2011).
Zhang, W. et al. Changes of angiotensin converting enzyme 2 in the occurrence and development of diabetic renal injury in rat. Sheng li xue bao:Acta physiologica Sinica 65(4), 402–408 (2013).
Verma, A. et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Molecular Therapy 20(1), 28–36 (2012).
Szabadfi, K. et al. Protective effects of the neuropeptide PACAP in diabetic retinopathy. Cell and tissue research 348(1), 37–46 (2012).
Dadras, F. & Khoshjou, F. NF-E2-related factor 2 and its role in diabetic nephropathy. Iranian journal of kidney diseases 7(5), 346 (2013).
Liu, Q. et al. Therapeutic effects of suppressors of cytokine signaling in diabetic nephropathy. Journal of Histochemistry & Cytochemistry 62(2), 119–128 (2014).
Papadimitriou, A. et al. Increase in AMPK brought about by cocoa is renoprotective in experimental diabetes mellitus by reducing NOX4/TGFβ-1 signaling. The Journal of nutritional biochemistry 25(7), 773–784 (2014).
Li, R. et al. Angelica injection promotes peripheral nerve structure and function recovery with increased expressions of nerve growth factor and brain derived neurotrophic factor in diabetic rats. Current neurovascular research 7(3), 213–222 (2010).
Yang, S. et al. Experimental study of the protective effects of SYVN1 against diabetic retinopathy. Scientific reports 5 (2015).
Perrone, L., Devi, T. S., Hosoya, K. I., Terasaki, T. & Singh, L. P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell death & disease 1(8), 65 (2010).
Atkinson, B. J., Griesel, B. A., King, C. D., Josey, M. A. & Olson, A. L. Moderate glut4 overexpression improves insulin sensitivity and fasting triglyceridemia in high-fat diet-fed transgenic mice. Diabetes 62, 2249–2258 (2013).
Shi, S. Y. et al. Adipocyte-specific deficiency of Janus kinase (JAK) 2 in mice impairs lipolysis and increases body weight, and leads to insulin resistance with ageing. Diabetologia 57, 1016–1026 (2014).
Araoka, T. et al. Transcription factor 7-like 2 (TCF7L2) regulates activin receptor-like kinase 1 (ALK1)/Smad1 pathway for development of diabetic nephropathy. Mol. Cells 30, 209–218 (2010).
Muendlein, A. et al. Single nucleotide polymorphisms of TCF7L2 are linked to diabetic coronary atherosclerosis. PLoS One 6 (2011).
Luo, J. et al. TCF7L2 variation and proliferative diabetic retinopathy. Diabetes 62, 2613–2617 (2013).
Miyata, T. et al. Increased pentosidine, an advanced glycation end product, in plasma and synovial fluid from patients with rheumatoid arthritis and its relation with inflammatory markers. Biochem Biophys Res Commun 244, 45–49 (1998).
Yousefzadeh, G., Pezeshki, S., Gholamhosseinian, A., Nazemzadeh, M. & Shokoohi, M. Plasma cystatin-C and risk of developing gestational diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 8, 33–35 (2014).
Li, L. et al. Plasma FGF-21 levels in type 2 diabetic patients with ketosis. Diabetes Res. Clin. Pract. 82, 209–213 (2008).
Mahmoud, R. A. K., el-Ezz, S. A. A. & Hegazy, A. S. Increased serum levels of interleukin-18 in patients with diabetic nephropathy. Ital. J. Biochem. 53, 73–81 (2004).
American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 29, 43–48 (2006).
Sindelar, D. K. et al. Low plasma leptin levels contribute to diabetic hyperphagia in rats. Diabetes 48(6), 1275–1280 (1999).
Sun, L. et al. Elevated plasma retinol-binding protein 4 is associated with increased risk of type 2 diabetes in middle-aged and elderly Chinese adults. J. Nutr. 144, 722–728 (2014).
Jguirim-Souissi, I. et al. Plasma Metalloproteinase-12 and Tissue Inhibitor of Metalloproteinase-1 Levels and Presence, Severity, and Outcome of Coronary Artery Disease. Am. J. Cardiol. 100, 23–27 (2007).
Thomsen, S. B., Rathcke, C. N., Zerahn, B. & Vestergaard, H. Increased levels of the calcification marker Matrix Gla Protein and the inflammatory markers YKL-40 and CRP in patients with type 2 diabetes and ischemic heart disease. Cardiovasc. Diabetol. 9, 7 (2010).
Li, Q. et al. Serum Angptl2 levels are independently associated with albuminuria in type 2 diabetes. Diabetes Res. Clin. Pract. 100, 385–390 (2013).
Makino, A. et al. High plasma levels of macrophage migration inhibitory factor are associated with adverse long-term outcome in patients with stable coronary artery disease and impaired glucose tolerance or type 2 diabetes mellitus. Atherosclerosis 213(2), 573–578 (2010).
Lim, H. S. et al. Plasma Vascular Endothelial Growth Factor, Angiopoietin-1, and Angiopoietin-2 in Diabetes Implications for cardiovascular risk and effects of multifactorial intervention. Diabetes Care 27(12), 2918–2924 (2004).
Tjeerdema, N. et al. Inflammation increases plasma angiopoietin-like protein 4 in patients with the metabolic syndrome and type 2 diabetes. BMJ open diabetes Res. care 2, e000034 (2014).
Jong, M. C. et al. Protection from obesity and insulin resistance in mice overexpressing human apolipoprotein C1. Diabetes 50, 2779–2785 (2001).
Kamoi, K., Ishibashi, M. & Yamaji, T. Thirst and plasma levels of vasopressin, angiotensin II and atrial natriuretic peptide in patients with non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 11, 195–202 (1991).
Yang, B., Hodgkinson, A. D., Shaw, N. A., Millward, B. A. & Demaine, A. G. Protective effect of statin therapy on connective tissue growth factor induction by diabetes in vivo and high glucose in vitro. Growth Factors 31, 199–208 (2013).
Wong, C. K. et al. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin. Exp. Immunol. 149, 123–131 (2007).
Németh, B. C. et al. Relevance of α-defensins (HNP1-3) and defensin β-1 in diabetes. World journal of gastroenterology: WJG 20(27), 9128 (2014).
Lee, I. J. et al. Growth arrest-specific gene 6 (Gas6) levels are elevated in patients with chronic renal failure. Nephrol. Dial. Transplant. 27, 4166–4172 (2012).
Müller, I. I. et al. Impact of counterbalance between macrophage migration inhibitory factor and its inhibitor Gremlin-1 in patients with coronary artery disease. Atherosclerosis 237, 426–432 (2014).
Zhang, X.-J. et al. Expression status of diabetes-associated genes in middle and aged cynomolgus monkeys. Zool. Res. 32, 300–6 (2011).
Narayanan, R. P. et al. IGFBP2 is a biomarker for predicting longitudinal deterioration in renal function in type 2 diabetes. Endocrine Connections 2(1), X1 (2013).
Bulló, M., Peeraully, M. R., Trayhurn, P., Folch, J. & Salas-Salvadó, J. Circulating nerve growth factor levels in relation to obesity and the metabolic syndrome in women. Eur. J. Endocrinol. 157, 303–10 (2007).
Bacon, S. et al. Serum levels of pancreatic stone protein (PSP)/reg1A as an indicator of beta-cell apoptosis suggest an increased apoptosis rate in hepatocyte nuclear factor 1 alpha (HNF1A-MODY) carriers from the third decade of life onward. BMC endocrine disorders 12(1), 13 (2012).
Yildirim, N. et al. The relationship between plasma MMP-9 and TIMP-2 levels and intraocular pressure elevation in diabetic patients after intravitreal triamcinolone injection. J. Glaucoma 17, 253–6 (2008).
(A.D.A.M. Medical Encyclopedia [Internet]. Atlanta (GA): A.D.A.M., Inc.; ©1997–2017. Nail abnormalities; [updated 2017 May 9]. Available at: https://medlineplus.gov/ency/article/000313.htm).
Rani, J., Shah, A.R. and Ramachandran, S. pubmed. mineR: An R package with text-mining algorithms to analyse PubMed abstracts. https://cran.r-project.org/web/packages/pubmed.mineR/index.html Journal of biosciences 40(4), 671–682 (2015).
Smith, G. K. limma: Linear Models for Microarray Data. Bioinforma. Comput. Biol. Solut. Using R Bioconductor 397–420, doi:citeulike-article-id:5722720 (2005).
Fellows, I. wordcloud: Word clouds. R package version 2 https://cran.r-project.org/web/packages/wordcloud/index.html Date of access: 09/09/2015 (2012).
Melville S. NCBI2R-An R package to navigate and annotate genes and SNPs https://github.com/cran/NCBI2R; Date of access: 15/07/2014 (2012).
Jensen, L. J., Saric, J. & Bork, P. Literature mining for the biologist: from information retrieval to biological discovery. Nat. Rev. Genet. 7, 119–29 (2006).
Barrett, T. et al. NCBI GEO: mining millions of expression profiles—database and tools https://www.ncbi.nlm.nih.gov/geo/ Nucleic acids research 33(suppl 1), D562–D566 Date of Access: 10/11/2015 (2005).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. Affy - Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Du, P., Kibbe, W. A. & Lin, S. M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008).
Iglewicz, B. and Hoaglin, D.C. How to detect and handle outliers (Vol. 16). Asq Press (1993).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Ferland-McCollough, D., Ozanne, S. E., Siddle, K., Willis, A. E. and Bushell, M. The involvement of microRNAs in Type 2 diabetes (2010).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003).
Merico, D., Isserlin, R., Stueker, O., Emili, A. & Bader, G. D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS One 5 (2010).
Stamm, S. et al. ASD: a bioinformatics resource on alternative splicing. Nucleic acids research 34(suppl 1), pp. D46–D55 (2006).
Matys, V. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–10 (2006).
Grillo, G. et al. UTRdb and UTRsite (RELEASE 2010): A collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 38 (2009).
Mandal, M. & Breaker, R. R. Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 5, 451–463 (2004).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic acids research 33(suppl 1), pp D121–D124 (2005).
Kin, T. et al. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic acids research. 35(suppl 1), D145–D148 (2007).
Macke, T. J. et al. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 29, 4724–4735 (2001).
Huang, H. Y., Chien, C. H., Jen, K. H. & Huang, H. D. RegRNA: An integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 34 (2006).
The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–12 (2015).
Acknowledgements
We thank Ms Priyanka Shrivastava and Ms Anasuya Bhargava for useful feedback. This work was supported by Council of Scientific and Industrial Research through grant BSC0122.
Author information
Authors and Affiliations
Contributions
The study was designed by S.R., I.M. and J.R. Data collection, organization and computation was done by J.R., I.M., A.P., N.S., A.M., M.V.R. PHP web enablement by J.R. Data analysis was performed by I.M., J.R., N.S., S.S. and B.L.P. Data reviews by S.R., I.M. and J.R. Manuscript written by J.R., N.S. and S.R. All authors have read and approved the manuscript for publication.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rani, J., Mittal, I., Pramanik, A. et al. T2DiACoD: A Gene Atlas of Type 2 Diabetes Mellitus Associated Complex Disorders. Sci Rep 7, 6892 (2017). https://doi.org/10.1038/s41598-017-07238-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07238-0
This article is cited by
-
Analysis of age-dependent gene-expression in human tissues for studying diabetes comorbidities
Scientific Reports (2023)
-
Prioritization of genes associated with type 2 diabetes mellitus for functional studies
Nature Reviews Endocrinology (2023)
-
Differential network analysis between sex of the genes related to comorbidities of type 2 mellitus diabetes
Applied Network Science (2023)
-
Ethnic disparities attributed to the manifestation in and response to type 2 diabetes: insights from metabolomics
Metabolomics (2022)
-
Identification of perturbed pathways rendering susceptibility to tuberculosis in type 2 diabetes mellitus patients using BioNSi simulation of integrated networks of implicated human genes
Journal of Biosciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
