
Abstract
Recent data highlight the presence, in HIV-1-seropositive patients with lymphoma, of p17 variants (vp17s) endowed with B-cell clonogenicity, suggesting a role of vp17s in lymphomagenesis. We investigated the mechanisms responsible for the functional disparity on B cells between a wild-type p17 (refp17) and a vp17 named S75X. Here, we show that a single Arginine (R) to Glycine (G) mutation at position 76 in the refp17 backbone (p17R76G), as in the S75X variant, is per se sufficient to confer a B-cell clonogenic potential to the viral protein and modulate, through activation of the PTEN/PI3K/Akt signaling pathway, different molecules involved in apoptosis inhibition (CASP-9, CASP-7, DFF-45, NPM, YWHAZ, Src, PAX2, MAPK8), cell cycle promotion and cancer progression (CDK1, CDK2, CDK8, CHEK1, CHEK2, GSK-3 beta, NPM, PAK1, PP2C-alpha). Moreover, the only R to G mutation at position 76 was found to strongly impact on protein folding and oligomerization by altering the hydrogen bond network. This generates a conformational shift in the p17 R76G mutant which enables a functional epitope(s), masked in refp17, to elicit B-cell growth-promoting signals after its interaction with a still unknown receptor(s). Our findings offer new opportunities to understand the molecular mechanisms accounting for the B-cell growth-promoting activity of vp17s.
Similar content being viewed by others


Introduction
HIV-1-associated lymphomas have not decreased after the introduction of combination antiretroviral therapy (cART) and non-Hodgkin’s lymphoma (NHL) represents the most common type of cancer1, 2 and the most frequent cause of death3, 4 in HIV-1-infected (HIV+) individuals. The HIV-1 genome is not integrated in the malignant B cells as seen for the oncogenic retrovirus HTLV-1. For this reason, the most shared assumptions rely on the indirect role of HIV-1 in lymphomagenesis. HIV-1-driven immune dysfunction with overproduction of B-cell stimulatory cytokines5 or loss of immune control, that promotes the reactivation of potentially oncogenic herpesviruses6, 7, are the most credited hypotheses up to date. However, novel findings support the possibility that HIV-1 may directly contribute to lymphomagenesis through mechanisms involving the biologic effects mediated by its gene products.
The HIV-1 matrix protein p17 (p17) is a 132 amino acid (aa)-long structural protein, composed of five major α-helixes and a highly basic platform consisting of three β strands8, 9. Four helixes are centrally organized to form a compact globular domain and a fifth helix (H5) in the COOH-terminus is slightly destabilized, due to the flexible C-terminal tail10, 11. The matrix protein is continuously released in the extracellular space from HIV-1-infected cells12. It has been detected in the plasma13 and in tissue specimens, such as brain14, liver15, bone marrow16 and lymph nodes of HIV+ patients17. In lymph nodes p17 accumulates and persists even during cART and in the absence of any HIV-1 replicative activity17, thus suggesting that it may be persistently expressed in the tissue microenvironment, even during pharmacological therapy, and promote chronic B-cell stimulation18, 19.
Latest data show that p17 expression in mice transgenic for a defective HIV-1 provirus is associated with lymphoma development20, 21. Moreover, p17 variants (vp17s), characterized by scattered mutation along the entire protein sequence22 or by specific aa insertions in the C-terminal region, display a potent B-cell growth-promoting activity triggering the PTEN/PI3K/Akt pathway22, 23, which is known to be crucial in lymphoma development24. Therefore, specific mutations within refp17 could induce a different pathogenetic potential to the viral protein. All these findings call for defining the structure-function relationship in clonogenic vp17s as compared to their wild-type counterpart.
In this study, we investigated the aa substitutions, the structural bases and the molecular mechanisms responsible for opposite effects in modulating B-cell growth between a vp17 derived from a Ugandan HIV-1 strain (subtype A1), named S75X22, and the wild type p17 (reference p17, refp17; from clone BH10 of the clade B isolate).
Here, we demonstrate that a single arginine (R) to glycine (G) mutation at position 76 in the refp17 backbone, as in S75X, is sufficient to induce dramatic changes in protein folding and stability, making p17 mutant capable of activating Akt and promoting B-cell proliferation.
Results
The R76G mutation in the refp17 backbone induces changes in the protein secondary structure and hydrogen bond network
We performed in silico studies to elucidate if aa mutations in S75X were responsible for changes in folding and stability of the viral protein as compared to refp17. Since mutated residues in S75X (Fig. 1A) are donors and acceptors of hydrogen bonds, we first performed long Molecular Dynamics simulation (MD) (500 ns) of refp17 to evaluate its hydrogen bond network. Side chains of mutated residues in S75X and involved in hydrogen bonds are shown in Fig. 1B. In order to identify key residues involved in interaction network, we evaluated hydrogen bonds during the entire refp17 MD and set the threshold of essential hydrogen bonds in the frames of all the trajectory to 75%25. The identified hydrogen bonds were E73-R76 (88%), N80-T84 (83%) and R58-E107 (81%) and the identification of E73-R76 as the most frequent hydrogen bond suggested us to further investigate its role in protein structure. In S75X the residue R76 results mutated in a G and this aa substitution leads to the loss of the E73-R76 hydrogen bond. Therefore, in order to investigate the role of this hydrogen bond in the stability and folding of the matrix protein, we modeled S75X and p17R76G, a p17 mutant with R76 replaced by a G, and performed long MD (500 ns).

Sequence and hydrogen bond network of refp17 and S75X. (A) Sequences are represented by the single-letter amino acid code. The amino acid sequence (aa 1–132) of the matrix protein clade B isolate BH10 p17 (UniProtKB P04585) was adopted as reference (refp17) for this analysis. Each amino acid residue of S75X not differing from refp17 sequence is represented by a dot. (B) At the top of the Figure, in the first row, the residues involved in the hydrogen bond network of p17 are shown and below the residues mutated in the variants S75X and p17R76G are indicated. The residues involved in hydrogen bond network are shown as sticks.
The stability of refp17, S75X and p17R76G was defined on the basis of these conditions along the dynamics: the total energy less than −40000 kcal/mol, RMSD of the structure in a range of 1 Angstrom around the centre of oscillations, the formation of hydrogen bonds, and the size of nonpolar surface area. Refp17 showed a higher stability than S75X and p17R76G, exhibiting a higher average number of hydrogen bonds (58 versus 48.5 and 55, respectively) and lower nonpolar surface areas (31.3% versus 33, 87% and 34, 23%, respectively).
In addition, the MD frames of p17s were clustered (% of the frames): two clusters (42% and 43%) in refp17, five clusters (29%, 26%, 15%, 9% and 7%) in the S75X and three clusters (25%, 23% and 22%) in p17R76G were obtained. The dispersion indices were 12.39, 31.65 and 20.00 for refp17, S75X and p17R76G, respectively. All these results suggest that S75X and p17R76G are less structured as compared to refp17.
In addition, using the DSSP software and calculating the average of helix propensities of each aa along the trajectory, we assessed whether mutations in S75X and p17R76G induce any changes in secondary structure26. Interestingly, S75X and p17R76G showed a disorganization in the N-terminal region of H3 and H4, and in the C-terminal region of H3 (Fig. 2). This was not observed in refp17, whose helices of globular domain were stable during simulation. A greater stability of H5 is observed in S75X and p17R76G as compared to refp17 (Fig. 2). The stability of H5 in S75X is due to the formation of the new hydrogen bond R91-E106 and in p17R76G of the R58-Q108 and Y86-E107 bonds. On the contrary, after 140 ns of MD in refp17 we observed a break in the middle of H5 (residue 105) resulting in displacement of structured C-terminal region towards the N-terminal region of H3. This shift allows the formation of a hydrogen bond R76-A112, obtaining a new interaction region. In all three proteins, fluctuations from helix to turn or coil structures were observed in the C-terminal region of H5 (Fig. 2).

Evolution of refp17, S75X and p17R76G protein structure by molecular dynamics simulations. (A) Time evolution of the secondary structural elements along the molecular dynamics simulation generated by DSSP. The X-axis represents the molecular dynamics trajectory time (in ns), while the residue numbers are shown on the Y-axis. (B) At the top of the Figure are indicated the residues involved in forming the helix structures of refp17 and determined experimentally by NMR (PDB code: 2HMX and 1TAM) and X-Ray (PDB code: 1HIW). In the graph the propensity for each amino acid residue to assume helix structure calculated during simulations of refp17 and its variants.
Overall, these findings show that changes in the hydrogen bond network, due to the insurgence of even a single point mutation (R76G), are responsible for changes in the secondary structure of refp17.
The R76G mutation destabilizes refp17 and alters its ability to oligomerize in solution
In order to confirm conformational shifts evidenced by MD, the structural conformation of refp17, S75X and p17R76G was investigated. To this aim we generated the three recombinant viral proteins and analyzed them in aqueous solution (each at 2.5 μM), using circular dichroism (CD) spectroscopy. All proteins displayed alpha-helical secondary structures at room temperature, as evidenced by a strong positive maximum at 195 nm and two negative minima at 208 and 222 nm (Fig. 3A), consistent with the known structural features of p178, 9. However, S75X and p17R76G were noticeably less helical than refp17, as indicated by the reduction in ellipticity, suggesting a partial unfold of proteins (Fig. 3A). In order to evaluate the impact of mutations on protein structure, we subjected refp17, S75X and p17R76G to heat-induced denaturation and monitored them at 222 nm by CD spectroscopy. As temperature was raised from 25 °C to 90 °C, all three proteins exhibited a co-operative unfolding with a single transition, typical of a compact globular protein (Fig. 3B). Data normalization based on a two-state protein denaturation model gave rise to a characteristic melting temperature (Tm at which 50% of protein is denatured or unfolded) for each protein. S75X and p17R76G had similar Tm values, approximately 10 °C lower than refp17 (Table 1). These results show that mutations in vp17s destabilize the matrix protein, corroborating findings from CD spectroscopy.

Structure and stability of refp17, S75X and p17R76G. (A) CD spectra of refp17, S75X, and p17R76G collected at room temperature at 2.5 μM in 10 mM phosphate buffer, pH 7.4. (B) Thermal denaturation of refp17, S75X, and p17R76G at 10 μM in PBS, pH 7.4, as monitored at 222 nm by CD spectroscopy. The experimental data were normalized according to a two-state protein denaturation model. Note that the thermal denaturation of p17 is irreversible due to protein aggregation. (C) Measurement of the size of folded refp17, S75X and p17R76G viral matrix proteins by the technique of dynamic light scattering at room temperature. (D) NMR spectra of refp17 (black), S75X (green), and p17R76G (red) showing their amide-amide NOEs.
Since temperature-induced unfolding is usually accompanied by protein aggregation27, we examined the ability of vp17s to oligomerize in solution by dynamic light scattering techniques (Table 1). In solution refp17 exists as monomer or trimer depending on protein concentration28. At 10 μM refp17 existed as a monomer (Fig. 3C), while S75X and p17R76G displayed a molecular weight 10 and 170 fold higher than refp17 (Fig. 3C), respectively. These data suggest that a single aa mutation occurring in the refp17 backbone can induce a massive self-association, indicating that some refp17 buried hydrophobic residues become exposed in S75X and p17R76G.
Then, we analyzed the three p17 proteins using 2-dimensional NMR spectroscopy and their amide-amide Nuclear Overhauser Effect Spectroscopy (NOESY) spectra. As shown in Fig. 3D, refp17 displayed a large number of well-dispersed amide-amide Nuclear Overhauser Effect (NOE) signals, which are characteristic of a well-folded alpha-helical protein. On the contrary, S75X and p17R76G showed very few amide-amide NOE signals in the same region of the spectrum, consistent with dampened signals arising from the formation of soluble aggregates.
Overall, our data suggest that a single R76G mutation in refp17 backbone can destabilize the viral protein with a reduction of alpha-helical secondary structures, thermal stability and soluble aggregates formation.
S75X and p17R76G promote B-cell growth
Destabilization of refp17 with changes in the secondary structure, induced by aa mutations, are known to confer conformational changes sufficient to endow the viral protein with B-cell growth-promoting activity23. Since in refp17 the E73-R76 hydrogen bond was identified by MD as an essential interaction for hydrogen bond network and CD and NMR spectroscopy confirm a destabilization of viral protein (Fig. 3), we aimed at determining whether p17R76G, a mutant bearing a G at position 76 instead of an R, as in S75X, could acquire a B-cell clonogenic activity. Then, we tested the p17R76G capability of modulating B-cell growth by soft agar clonogenic assays. As shown in Fig. 4A, refp17 inhibited the colony-forming ability of Raji B cells as compared to untreated cultures, while S75X and p17R76G enhanced their clonogenic activity. These results confirm that the rupture of the E73-R76 hydrogen bond generates significant structural changes conferring to the matrix protein a S75X-like B-cell clonogenic activity.

Effect of refp17, S75X and p17R76G on B-cell activity. (A) Raji were plated in twelve-well plates and, after four days, medium was replaced by fresh medium with the indicated concentration of refp17, S75X and p17R76G. Cells not treated (NT) were used as negative control. The cell growth was analyzed by using MTT. Data represent the average number of colonies ± SD from three independent experiments performed in triplicate. The statistical significance between control and treated cultures was calculated using one-way ANOVA performed separately for each concentration of p17 variants and Bonferroni’s post-test was used to compare data; ***P < 0.001. (B–D) Cells were treated for 5 min with 0.05, 0.1, 0.5 μg/ml of refp17 (B), S75X (C) and p17R76G (D). Untreated cells were used as control. Western blot analysis of Raji lysates shows that refp17 inhibits the activation of Akt and maintains PTEN in an active state (B), as shown by the respective phosphorylation state at any concentration tested, verified by densitometric analysis and plotting of the pAkt/Akt and pPTEN/GAPDH. On the contrary, either S75X or p17R76G induce the activation of Akt and maintains PTEN in an inactive state (C,D), as shown by the increased phosphorylation, verified by densitometric analysis and plotting of the pAkt/Akt and pPTEN/GAPDH. In the left panel blots from one representative experiment of three with similar results are shown. In the right panels, values reported for phosphorylation of Akt and PTEN are the mean ± SD of three independent experiments. Statistical analysis was performed by one-way ANOVA and the Bonferroni’s post-test was used to compare data; *P < 0.05; **P < 0.01; ***P < 0.001.
S75X and p17R76G activate the PTEN/PI3K/Akt pathway in B cells
The PTEN/PI3K/Akt pathway represents the major intracellular signaling cascade known to regulate proliferation and malignant transformation29, 30. Refp17 was shown to enhance PTEN activity and down-modulate the PI3K/Akt signaling pathway, exerting an anti-proliferative effect on B cells, while S75X was shown to block PTEN and activate Akt, thus promoting B-cell growth22. Due to clonogenic activity of p17R76G (Fig. 4A), we explored its capability to modulate the PTEN/PI3K/Akt pathway. As shown in Fig. 4B, Raji cells stimulated with refp17 showed a significant Akt inhibition, as expected, while S75X and p17R76G phosphorylated Akt kinase (Fig. 4C and D). Since Akt can be modulated by PTEN30, we also evaluate the Ser/Thr phosphorylated PTEN inactivation form. As shown in Fig. 4C and D, cells treated with S75X and p17R76G showed an increased PTEN phosphorylation as compared to unstimulated cells, whereas treatment of cells with refp17 resulted in a reduced PTEN phosphorylation level (Fig. 4B). Overall, these data show that p17R76G, like S75X, activates the proliferative PI3K/Akt pathway in B cells through PTEN down-modulation.
S75X and p17R76G modulate different signaling molecules involved in cell cycle regulation
Assessment of protein phosphorylations is essential for understanding intracellular activities controlling cell functions. To further investigate the intracellular mechanisms involved in the B-cell growth-promoting activity of S75X and p17R76G, we assessed how these proteins modulate intracellular phosphorylations by employing a high-throughput antibody array technology. The lysates of Raji cells, stimulated or not with refp17, p17R76G or S75X (0.1 μg/ml) were processed by Kinexus Bioinformatics Corporation for Kinex™ KAM-850 Antibody Microarray. Then, in order to rule out false positive array signals, all the protein modulated by S75X and p17R76G were confirmed by a multi-immunoblotting analysis performed by Kinexus. The signals relative to the 23 proteins modulated by S75X and p17R76G, verified by densitometric analysis and normalized to refp17, are reported in Table 2. Literature data mining and GO biological process database highlighted that most of modulated proteins, as expected, are involved in cell cycle regulation (CDK1, CDK2, CDK8, CHEK1, CHEK2, GSK-3 beta, NPM, PAK1, PP2C-alpha) and apoptosis (CASP-9, CASP-7, DFF-45, NPM, YWHAZ, Src, PAX2, MAPK8).
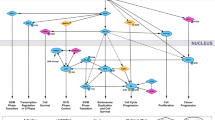
In Fig. 5 we established the known and experimentally verified molecular interactions among the proteins reported in Table 2 using STRING database31 of Protein-Protein Interaction Network and literature data mining. Those molecules not displaying confirmed interactions (CaMKK2, CDK8, Lyn, PP2C-delta, SOCS-4, Src, CHEK1, GSK-3 beta, PP5, PKC-beta) were kept out from the signaling network, even if we cannot completely rule out their involvement in vp17s-induced B-cell growth. As shown in Fig. 5, S75X and p17R76G trigger the PI3K/Akt signaling pathway, which in turn could activate MAPK832 and YWHAZ33, thus creating a positive feedback loop further strengthening Akt activation34. In addition, MAPK8, besides its ability to activate PAX2 and promote cancer cell proliferation35, can trigger YWHAZ and inhibit DNA fragmentation process during apoptosis by interacting with DFF-4536. PP2C-alpha up-regulation can also concurs likely to further activate PI3K/Akt pathway37. Moreover, Akt activation can modulate the activity of PAK138, 39, CASP-940, DFF-4536, 41, NPM42, 43, CDK1 and CDK244, 45. Among these factors, a central role can be exerted by NPM, which is known to be directly activated by Akt, but also by YWHAZ and CDK1/246,47,48. Finally, CHEK2 activation can be involved in cytoprotection and cell survival49.

Representation of the putative signaling pathways involved in B-cell clonogenicity induced by interaction of S75X/p17R76G with p17R(s). Stimulation of B cells with the clonogenic p17 proteins, S75X and p17R76G, induces the activation of several molecules involved in promoting cell survival, cell cycle progression and in inhibiting apoptosis, and STRING database and literature data mining were used to identify known and experimentally verified interactions. The several kinases involved in the pathway are indicated by orange diamonds, the other proteins are represented by light red ellipses. PI3K: phosphatidylinositol-3-kinase; Akt: Protein kinase B;PP2C-alpha: Protein phosphatase 1A; MAPK8: Mitogen-activated protein kinase 8; YWHAZ: 14-3-3 protein zeta/delta; CHEK2: Checkpoint kinase 2; CDK1: Cyclin-dependent kinase 1; CDK2: Cyclin-dependent kinase 2; NPM: Nucleophosmin; DFF-45: DNA fragmentation factor subunit alpha; CASP-9: caspase-9; CASP-7: caspase-7; PAK1: Serine/threonine-protein kinase PAK 1; PAX2: Paired box protein Pax-2.
These data highlight that the activation of PI3K/Akt pathway in B cells by clonogenic S75X and p17R76G can orchestrate the function of several molecules involved in promoting tumor survival and proliferation.
Discussion
The function of a globular protein is governed by its 3D structure, while different biological functions can be performed by a protein that lacks the ordered secondary structure and thermal stability50,51,52. As several cancer-associated proteins, p17 possesses high levels of predicted intrinsic disorders53, 54, which subtend to a partially unfolded status of specific regions. Previously, we showed that in S75X, which contains aa mutations scattered throughout the p17 protein sequence, specific hydrogen bonds stabilizing the refp17 structure are lost, giving rise to a partially unfolded - B-cell growth-promoting - protein18. However, it is known that different aa residues can be mutated in vp17s without loss or change in their refp17-like functional activity, suggesting that the clonogenic activity of vp17s do not depend from protein sequence, but only from specific mutations capable of endowing p17s with a B-cell growth-promoting activity23. MD simulations showed that S75X displays a single aa mutation (R76G) that is likely to be the most responsible for protein destabilization, with reduction of alpha-helical secondary structures and for shifting the equilibrium toward a partially unfolded conformation. CD and 2-dimensional NMR spectroscopy confirmed this prediction using a recombinant p17 mutant carrying the single R76G replacement in the refp17 backbone (p17R76G). As expected, clonogenic assays showed that the replacement of R to G at position 76, as in S75X, is sufficient to endow refp17 with a B cells growth-promoting activity. Therefore, aa mutations, which alter the hydrogen bond network and contribute to an even partial unfolding process of refp17, confer to vp17s different functional outcomes compared to refp17.
Unfolding and subsequent self-assembly of proteins into various aggregates are common molecular mechanisms involved in important human diseases55, 56. Our data show that the partially unfolded conformational ensemble of vp17s favors specific intermolecular interactions and protein aggregation. Indeed, vp17s exhibit higher nonpolar surface areas than refp17, probably due to the exposure of some buried hydrophobic residues, which induces them to massively self-aggregate. Further studies will address if some of the p17 cell toxic activities57 are dependent on this aggregation status.
The p17R76G protein, similarly to S75X22, was also found to activate Akt kinase, modulating PTEN activity. High-throughput phosphoproteomic and bioinformatics analysis highlighted a central role of Akt kinase in the proliferative signaling pathways induced by S75X and p17R76G in B cells. Indeed, Akt can modulate the function of several molecules as PP2C-alpha, YWHAZ, MAPK8, PAX2, PAK1, CASP-9, CASP-7, DFF-45, NPM, PAX2, CDK1, CDK2, CHEK2, involved in apoptosis inhibiton, cell cycle promotion and cancer progression. All these findings were not unexpected since the PTEN/PI3K/Akt pathway represents the major signaling cascade known to play a central role in tumorigenesis58, 59.
All molecules modulated by S75X and p17R76G, used as input for the Cytoscape ClueGO functional analysis tool60 to cluster them according to the KEGG biological pathway, result to be related to several known biological pathways. As shown in Fig. 6, most of the identified signaling pathways are related to cancer development and progression, as PI3K/Akt, p53, MAPK and ErbB. It is important to underline that molecules modulated by S75X and p17R76G are also involved in signaling triggered by oncogenic viruses, like EBV, HTLV-1 and HBV. This latter observation suggests that different viruses, and among them HIV-1, may exploit common paths to promote cell proliferation. Moreover, EBV infection was recently shown to up-regulate p17Rs expression on primary B lymphocytes, thereby making these cells permissive to the biological effects mediated by p17s19. Interestingly, molecules modulated by S75X and p17R76G were also related to the prolactin signaling pathway, supporting previous data showing the presence of higher serum prolactin in HIV+ patients than in healthy individuals and the capability of prolactin to influence normal lymphocyte mitogenesis and lymphoid tumor growth61,62,63. Finally, some molecules modulated by clonogenic vp17s result to be related to chemokine signaling pathway and this finding is not surprising, since p17 is known to act as a viral chemokine15, 64,65,66.

Representation of KEGG pathways significantly regulated by S75X and p17R76G. The proteins modulated by p17s were used as input for the Cytoscape ClueGO functional analysis tool to cluster the proteins according to the KEGG pathways database. The proteins are represented by yellow circles. The pathways are represented by red circles and the circle size is directly proportional to statistical relevance (3 * 10−3 > p < 8 * 10−6). An edge is present when the protein is involved in the pathway.
Recently, we showed that vp17s derived from HIV+ NHL patients were characterized by specific aa insertions in the C-terminal region, tempting to ascribe this activity to the C-terminus of the protein interacting directly with its putative receptors23. Nevertheless, we previously described that the presence of H5 in p17 is not necessary for protein clonogenic activity22, highlighting the intrinsic capability of p17 to exert B-cell clonogenic activity following protein unfolding. Indeed, truncation of the last 36 aa residues of p17 resulted in a protein (p17Δ36) capable of promoting B-cell clonogenicity through PTEN/PI3K/Akt pathway. Moreover, the different biological activity of S75X and p17R76G compared to refp17, underlines how the matrix protein binding to and signaling through p17R(s) can potentially involve distinct determinants of structure or conformations. For these reasons, we hypothesize that a specific clonogenic epitope responsible for B-cell clonogenic activity, located probably in N-terminal or in the core of matrix protein, is masked in the properly folded monomeric p17, but exposed in misfolded, aggregated B-cell vp17s. Therefore, the molecular reasons for the opposite mechanisms among mutated proteins and wild-type p17 may depend on the exposure or not of a clonogenic epitope and from the activation of the PI3K/Akt pathway, which is specific for the mutated clonogenic proteins. This downstream signaling pathway might be ascribed to the interaction of the viral mutated proteins, in their unfolded or aggregated form, with a different p17R(s) from the ones known engaged by refp17. Up to date, the direct interactor of clonogenic mutated proteins is still unknown and its identification will be sure matter of further investigations.
In conclusion, our study represents the first molecular and structural characterization of a single point mutated p17 displaying B-cell clonogenic activity. Our findings open the way for a better understanding of the structure-function relationship underlying the B-cell clonogenic activity of some vp17s. Moreover, demonstration that clonogenic vp17s exert their action on B cells through activation of the PTEN/PI3K/Akt pathway, which represents a critical driver of lymphoma development and metastasis, may offer new opportunities to identify novel treatment strategies in combating HIV-related B-cell lymphoma. Finally, our results provide the rational background for further studies aimed to assess the possible molecular signatures identifying vp17s with B-cell clonogenic activity and recognition of HIV+ individuals at risk to develop lymphoma.
Materials and Methods
Cell cultures
Human lymphoblastoid cell line (Raji) was obtained from the American Type Culture Collection (ATCC) and cultured in RPMI-1640 medium containing 10% fetal calf serum, 1 mM L-glutamine and 1mM sodium pyruvate.
Recombinant proteins
Purified endotoxin-free recombinant HIV-1 matrix protein p17, S75X and p17R76G were produced as previously described22. The mutated matrix protein p17R76G, in which the arginine in position 76 was replaced by a glycine, was generated from p17 sequence by using Quick Change Site-directed mutagenesis Kit (Stratagene).
Molecular Modeling
The structures of refp17 were obtained from the Protein Data Bank (PDB) code 1TAM, whereas homology modeling (Modeler) was used to obtain the structures of S75X and p17R76G. The PDB code 1TAM, which corresponds to the refp17 studied experimentally, was used as a template. The models were optimized with AMBER program67 and checked using PROCHECK68.
Molecular Dynamics (MD)
MD simulations were performed with AMBER 12 software package67 and AMBER ff99SB force field. The system was solvated in a truncated octahedron periodic box by using TIP3P and counter ions were added to neutralize the system. The minimizations were performed keeping the position of protein restrained for tree minimization steps, by a force constant of 5000 Kcal/mol/Ų in the first, 100 Kcal/mol/Ų in the second, 10 Kcal/mol/Ų in the third and without protein restrained in the fourth. Next, the system was heated to 300 K using position restraints on entire protein with force constant of 10 kcal/mol/Ų, from 0 K to 100 K at constant volume in 3 ns, from 100 to 300 at constant pressure in 2 ns.
Bond lengths involving hydrogens were constrained using the SHAKE algorithm and the equations of motion were integrated with a 2-fs time step. The non-bonded cutoff distance was 8Å and the Particle Mesh Ewald method was used to calculate long-range electrostatics interactions. The temperature of the system was regulated using the Langevin thermostat. Equilibration was reached in constant NPT ensemble for 7 ns. After equilibration, production MD was performed at 300 K using constant pressure without restraints for 500 ns. DSSP program was used to assign secondary structures26.
The convergence was evaluated calculating the time point beyond which the RMSD of the structure stayed in a range of 1 Angstrom around the centre of oscillations. The RMSD was calculated considering only the alpha carbon in the not flexible regions (RMSF < 3 Angstrom).
To assess the different conformations of p17s we performed cluster analysis using the default hierarchical agglomerative algorithm implemented in the cpptraj tool (AmberTools). The distance matrix was computed using RMS distance between each pair of frame. The dispersion of conformations along the dynamics within of the clusters was evaluated considering a dispersion index that takes into account the fraction of frames (FR i ) and the average of RMSD within each cluster: \({\sum }_{i=1}^{Nc}RMS{D}_{i}\cdot (1-F{R}_{i})\), where N c is the number of considered clusters.
The non-polar surfaces area was calculated using the software MDTraj69.
CD spectroscopy and thermal denaturation
CD spectra of p17s at 2.5 μM in 10 mM PBS (pH 7.4) were obtained at room temperature on a Jasco J-810 spectropolarimeter using a 1-mm quartz cuvette. Protein thermal denaturation was carried out in PBS on the Jasco spectropolarimeter equipped with a temperature controller. 2.5 mL of protein solution (approximately 10 μM) prepared in PBS (pH 7.4) was aliquoted into a 3 mL cuvette. Under constant stirring, CD measurements at 222 nm were made at one degree interval between 25 °C and 90 °C, at a heating rate of 1 °C per minute. After each 1-minute heating, the solution in the cuvette waited for 20 s before signals were sampled over a 16 sec period. Heating and data acquisition were fully automated with control software provided by Jasco. Denaturation data were fitted to the following six-parameter equation derived from a two-state protein denaturation model as previously described23, 70, 71:
where Tm is melting temperature at which 50% of protein is denatured, ∆Hm is the enthalpy of denaturation, αn1, αn2, αd1, and αd2 are parameters for the parabolic equations defining native and denatured states, and the gas constant R = 8.314 J mol−1 K−1. The experiments for each protein were repeated three times.
Dynamic light scattering
Concentrated protein solutions were filtered using a Whatman Anotop 10 filter (0.22 μm) and, after UV quantification, diluted in PBS to a final concentration of 10 μM each. Protein size measurements were carried out at 25 °C, pH 7.4, with a total ionic strength of 0.1 M, on a Malvern Zetasizer Nano ZS instrument. Twenty runs of 30 s each were taken and averaged, and the data were processed using the manufacturer provided software.
NMR spectroscopy
NMR spectra were recorded at 25 °C on an 800 MHz (800.27 MHz for protons) Bruker Avance-series NMR spectrometer equipped with four frequency channels and a 5 mm triple-resonance z-axis gradient cryogenic probehead. A one-second relaxation delay was used and quadrature detection in the indirect dimensions was obtained with states-TPPI phase cycling; initial delays in the indirect dimensions were set to give zero- and first-order phase corrections of 90° and −180°, respectively72, 73. Data were processed using the processing program nmrPipe on Mac OS X workstations74. 2D NOESY experiments with a 150 ms mixing time were collected to monitor changes in the backbone and side-chain 1H protein resonances75. Typical NMR samples contained 3 mg/ml protein in a 10 mM sodium phosphate and 25 mM sodium chloride buffer (pH 7.4) to which 10% D2O (v/v) was added.
Soft agar anchorage-independent growth assay
Raji cell clonogenic activity was evaluated in soft agar assays as previously described18, 22, 23.
Western blot, high throughput phosphoproteomics and bioinformatics analysis
Raji cells, starved for 24 hours by serum deprivation, were stimulated or not with refp17, p17R76G and S75X (0.05, 0.1, 0.5 μg/ml) for 5 min, then lysed and prepared for KinexTM Antibody Microarray (KAM-850) according to manufacturer’s instructions (Kinexus Bioinformatics Corporation). Then, the samples were checked for pAkt and pPTEN levels by western blot analysis, as previously described23. In particular, the blots were incubated overnight at 4 °C with 1) mAb to pAkt (Cell Signaling Technology), 2) mAb to total Akt (Cell Signaling Technology), 3) mAb pPTEN (Ser380/Thr382/383) (Cell Signaling Technology), 3) mAb to GAPDH (glyceraldehyde-3-phosphate dehydrogenase). The lysates of cells stimulated with refp17, p17R76G and S75X (0.1 μg/ml) for 5 min were sent to Kinexus Bioinformatics Corporation for processing by Kinex™ KAM-850 Antibody Microarray, which contains 517 pan-specific and 337 phoshpo-site-specific probes, which recognize different epitopes on 466 proteins. The raw data, processed by Kinexus with the ImaGene software, were filtered according to company criteria. Only probes matching these criteria where considered for further analysis: signal to noise ratio >1.6; or signal to noise ratio <1.6 and noise inferior than the average noise of the array; maximum variation coefficient <0.2. The analysis was led with custom R, Visual Basic and MySQL procedures. Z-scores for normalization and z-ratios for comparisons were calculated as previously described76, according to the manufacturer’s recommendations, and a significativity threshold of 1.1 was adopted for the z-ratios. In order to rule out false positive array signals, a subsequent multi-immunoblotting verification step was performed according to the manufacturer’s recommendations. Therefore, on the 54 most relevant selected probes a pre-screening step was performed by Kinexus to select the reactive antibodies and rule out the false positive array signals due to antibody cross-reactivity. After pre-screening, 36 selected antibodies were used for a multi-immunoblotting analysis by Kinexus. Blot raw quantification data were processed by log transformation and normalization to either not treated or p17 treated samples for the interpretation of the results. STRING database was used to identify known and experimentally verified molecular interactions among proteins modulated by p17s31. Literature data mining was also performed by PubMed (http://www.ncbi.nlm.nih.gov/pubmed), PhosphoSite (http://www.phosphosite.org/), Uniprot (http://www.uniprot.org) and Genecards (http://www.genecards.org).
The modulated proteins were also used as input for the Cytoscape ClueGO functional analysis tool50 to cluster the proteins according to the KEGG pathways database.
Statistical analysis
Data obtained from multiple independent experiments are expressed as the means ± the standard deviations (SD). The data were analyzed for statistical significance using one-way ANOVA. Bonferroni’s post-test was used to compare data. Differences were considered significant at P < 0.05. Statistical tests were performed using Prism 5 software (GraphPad).
References
Shiels, M. S. et al. Cancer burden in the HIV-infected population in the United States. J. Natl. Cancer Inst. 103, 753–762 (2011).
Simard, E. P., Pfeiffer, R. M. & Engels, E. A. Cumulative incidence of cancer among individuals with acquired immunodeficiency syndrome in the United States. Cancer 117, 1089–1096 (2011).
Carbone, A. et al. Diagnosis and management of lymphomas and other cancers in HIV-infected patients. Nat. Rev. Clin. Oncol. 11, 223–238 (2014).
Vandenhende, M. A. et al. Cancer-Related Causes of Death among HIV-Infected Patients in France in 2010: Evolution since 2000. PLoS One 10, e0129550 (2015).
Epeldegui, M., Vendrame, E. & Martínez-Maza, O. HIV-associated immune dysfunction and viral infection: role in the pathogenesis of AIDS-related lymphoma. Immunol. Res. 48, 72–83 (2010).
da Silva, S. R. & de Oliveira, D. E. HIV, EBV and KSHV: viral cooperation in the pathogenesis of human malignancies. Cancer Lett. 305, 175–185 (2011).
Dolcetti, R., Gloghini, A., Caruso, A. & Carbone, A. A lymphomagenic role for HIV beyond immune suppression? Blood 127, 1403–1409 (2016).
Massiah, M. A. et al. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 244, 198–223 (1994).
Hill, C. P., Worthylake, D., Bancroft, D. P., Christensen, A. M. & Sundquist, W. I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci USA 93, 3099–3104 (1996).
Verli, H., Calazans, A., Brindeiro, R., Tanuri, A. & Guimarães, J. A. Molecular dynamics analysis of HIV-1 matrix protein: clarifying differences between crystallographic and solution structures. J. Mol. Graph. Model 26, 62–68 (2007).
Ohori, Y. et al. Flexible and rigid structures in HIV-1 p17 matrix protein monitored by relaxation and amide proton exchange with NMR. Biochim. Biophys. Acta 1844, 520–526 (2014).
Caccuri, F. et al. Cellular aspartyl proteases promote the unconventional secretion of biologically active HIV-1 matrix protein p17. Sci. Rep. 6, 38027 (2016).
Fiorentini, S. et al. HIV-1 matrix protein p17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc. Natl. Acad. Sci. USA 105, 3867–3872 (2008).
Budka, H. Human immunodeficiency virus (HIV) envelope and core proteins in CNS tissues of patients with the acquired immune deficiency syndrome (AIDS). Acta neuropathol. 79, 611–619 (1990).
Caccuri, F. et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc. Natl. Acad. Sci. USA 109, 14580–14585 (2012).
Wiley, E. L. & Nightingale, S. D. Opportunistic events and p17 expression in the bone marrow of human immunodeficiency virus-infected patients. J. Infect. Dis. 169, 617–620 (1994).
Popovic, M. et al. Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 102, 14807–14812 (2005).
Caccuri, F. et al. Simian immunodeficiency virus and human immunodeficiency virus type 1 matrix proteins specify different capabilities to modulate B-cell growth. J. Virol. 88, 5706–5717 (2014).
Martorelli, D. et al. A natural HIV p17 protein variant up-regulates the LMP-1 EBV oncoprotein and promotes the growth of EBV-infected B-lymphocytes: implications for EBV-driven lymphomagenesis in the HIV setting. Int. J. Cancer 137, 1374–1385 (2015).
Curreli, S. et al. B cell lymphoma in HIV transgenic mice. Retrovirology 10, 92 (2013).
Carroll, V. A. et al. Expression of HIV-1 matrix protein p17 and association with B-cell lymphoma in HIV-1 transgenic mice. Proc. Natl. Acad. Sci. USA 113, 13168–13173 (2016).
Giagulli, C. et al. Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B-cells. PLoS One 6, e17831 (2011).
Dolcetti, R. et al. Role of HIV-1 matrix protein p17 variants in lymphoma pathogenesis. Proc. Natl. Acad. Sci. USA 112, 14331–14336 (2015).
Jiang, B. H. & Liu, L. Z. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim Biophys. Acta 1784, 150–158 (2008).
Durrant, J. D. & McCammon, J. A. HBonanza: a computer algorithm for molecular-dynamics-trajectory hydrogen-bond analysis. J. Mol. Graph. Model 31, 5–9 (2011).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Benjwal, S., Verma, S., Röhm, K. H. & Gursky, O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 15, 635–639 (2006).
Massiah, M. A. et al. Comparison of the NMR and X-ray structures of the HIV-1 matrix protein: evidence for conformational changes during viral assembly. Protein Sci 5, 2391–2398 (1996).
Adjei, A. A. & Hidalgo, M. Intracellular signal transduction pathway proteins as targets for cancer therapy. J. Clin. Oncol. 23, 5386–5403 (2005).
Cantley, L. C. & Neel, B. G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 96, 4240–4245 (1999).
von Mering, C. et al. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 (2003).
Hartman, A. D. et al. Constitutive c-jun N-terminal kinase activity in acute myeloid leukemia derives from Flt3 and affects survival and proliferation. Exp. Hematol. 34, 1360–1376 (2006).
Yoshida, K., Yamaguchi, T., Natsume, T., Kufe, D. & Miki, Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell. Biol. 7, 278–285 (2005).
Neal, C. L. et al. Overexpression of 14-3-3ζ in cancer cells activates PI3K via binding the p85 regulatory subunit. Oncogene 31, 897–906 (2012).
Cai, Y. et al. Phosphorylation of Pax2 by the c-Jun N-terminal kinase and enhanced Pax2-dependent transcription activation. J. Biol. Chem. 277, 1217–1222 (2002).
Ewing, R. M. et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 3, 89 (2007).
Yoshizaki, T. et al. Protein phosphatase-2C alpha as a positive regulator of insulin sensitivity through direct activation of phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J Biol. Chem. 279, 22715–2272 (2004).
Banerjee, M., Worth, D., Prowse, D. M. & Nikolic, M. Pak1 phosphorylation on T212 affects microtubules in cells undergoing mitosis. Current Biology 12, 1233–1239 (2002).
Radu, M., Semenova, G., Kosoff, R. & Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 14, 13–25 (2014).
Xu, D. et al. Apoptotic block in colon cancer cells may be rectified by lentivirus mediated overexpression of caspase-9. Acta Gastroenterol. Belg. 76, 372–380 (2013).
Jin, J. et al. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 14, 1436–1450 (2004).
Lee, S. B. et al. Nuclear Akt interacts with B23/NPM and protects it from proteolytic cleavage, enhancing cell survival. Proc. Natl. Acad. Sci. USA 105, 16584–16589 (2008).
Koike, A. et al. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 70, 6746–6756 (2010).
Shtivelman, E., Sussman, J. & Stokoe, D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr. Biol. 12, 919–924 (2002).
King, F. W., Skeen, J., Hay, N. & Shtivelman, E. Inhibition of Chk1 by activated PKB/Akt. Cell Cycle 3, 634–637 (2004).
Tokuyama, Y., Horn, H. F., Kawamura, K., Tarapore, P. & Fukasawa, K. Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J. Biol. Chem. 276, 21529–21537 (2001).
Negi, S. S. & Olson, M. O. Effects of interphase and mitotic phosphorylation on the mobility and location of nucleolar protein B23. J. Cell. Sci. 119, 3676–3685 (2006).
Pozuelo-Rubio, M. Proteomic and biochemical analysis of 14-3-3-binding proteins during C2-ceramide-induced apoptosis. FEBS J. 277, 3321–3342 (2010).
Ashwell, S. & Zabludoff, S. DNA damage detection and repair pathways–recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 14, 4032–4037 (2008).
Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M. & Obradović, Z. Intrinsic disorder and protein function. Biochemistry 41, 6573–6582 (2002).
Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005).
Uversky, V. N., Oldfield, C. J. & Dunker, A. K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384 (2005).
Goh, G. K., Dunker, A. K. & Uversky, V. N. A comparative analysis of viral matrix proteins using disorder predictors. Virol. J. 5, 126 (2008).
Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradovic, Z. & Dunker, A. K. Intrinsic disorder in cell-signaling and cancer- associated proteins. J. Mol. Biol. 323, 573–584 (2002).
Stefani, M. & Dobson, C. M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699 (2003).
Winklhofer, K. F., Tatzelt, J. & Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336–349 (2008).
Fiorentini, S., Marini, E., Caracciolo, S. & Caruso, A. Functions of the HIV-1 matrix protein p17. New Microbiol. 29, 1–10 (2006).
Li, L. et al. The emering role of the PI3-K-Akt pathway in prostate cancer progression. Prostate Cancer Prostatic Dis. 8, 108–118 (2005).
McCubrey, J. A. et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 46, 249–279 (2006).
Bindea, G. et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Matera, L. et al. Expression of prolactin and prolactin receptors by non-Hodgkin’s lymphoma cells. Int. J. Cancer 85, 124–130 (2000).
Montero, A., Bottasso, O. A., Luraghi, M. R., Giovannoni, A. G. & Sen, L. Association between high serum prolactin levels and concomitant infections in HIV-infected patients. Hum. Immunol. 62, 191–196 (2001).
Ram, S., Acharya, S., Fernando, J. J., Anderson, N. R. & Gama, R. Serum prolactin in human immunodeficiency virus infection. Clin. Lab. 50, 617–620 (2004).
Giagulli, C. et al. HIV-1 matrix protein p17 binds to the IL-8 receptor CXCR1 and shows IL-8-like chemokine activity on monocytes through Rho/ROCK activation. Blood 119, 2274–2283 (2012).
Caccuri, F. et al. HIV-1 matrix protein p17 promotes lymphangiogenesis and activates the endothelin-1/endothelin B receptor axis. Arterioscler. Thromb. Vasc. Biol. 34, 846–856 (2014).
Caccuri, F., Marsico, S., Fiorentini, S., Caruso, A. & Giagulli, C. HIV-1 Matrix Protein p17 and its Receptors. Curr. Drug Targets 17, 23–32 (2016).
Case, D. A. et al. AMBER 12, University of California, San Francisco (2012).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993).
McGibbon, R. T., Schwantes, C. R. & Pande, V. S. Statistical model selection for markov models of biomolecular dynamics. J. Phys. Chem. B. 118, 6475–6481 (2014).
Wu, Z. et al. Total chemical synthesis of N-myristoylated HIV-1 matrix protein p17: structural and mechanistic implications of p17 myristoylation. Proc. Natl. Acad. Sci. USA 101, 11587–11592 (2004).
Pace, C. N. & Scholtz J. M. Protein structure: A Practical Approach, (ed. Creighton T. E.) 299–321 (IRL press, Oxford 1997).
Marion, D. et al. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin 1 beta. Biochemistry 28, 6150–6156 (1989).
Bax, A. & Ikura, M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J. Biomol. NMR 1, 99–104 (1991).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Mori, S., Abeygunawardana, C., Johnson, M. O. & van Zijl, P. C. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B. 108, 94–98 (1995).
Cheadle, C., Cho-Chung, Y. S., Becker, K. G. & Vawter, M. P. Application of z-score transformation to Affymetrix data. Appl Bioinformatics 2, 209–217 (2003).
Acknowledgements
This work was supported by Italian Association for Cancer Research (AIRC, IG16797 to C.L. and IG14287 to R.D.), Italian Ministry of Education, University and Research (MIUR, PRIN 2009 to C.L., PRIN 2015 to L.M.), Flagship “InterOmics” Project (PB05 to L.M.), Nanomedicine project University of Verona and Fondazione Cariverona to C.L. The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Contribution: C.G. and A.C. conceived the study, designed experiments and wrote the manuscript. F.C. and S.M. performed and analyzed data on clonogenic assays; C.G., S.Z., B.O. and C.L. analyzed phosphoarray data and performed bioinformatics analysis; P.D., A.O. and L.M. performed molecular modeling and dynamics; W.H., K.V. and W.L. performed and analyzed protein folding and aggregation; C.G., S.F. and R.D. performed and analysed data on signaling pathways triggered by refp17 and vp17s. All the authors contributed to the interpretation of the results, gave relevant input to the intellectual content of the manuscript, revised it, and approved its final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Giagulli, C., D’Ursi, P., He, W. et al. A single amino acid substitution confers B-cell clonogenic activity to the HIV-1 matrix protein p17. Sci Rep 7, 6555 (2017). https://doi.org/10.1038/s41598-017-06848-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06848-y
This article is cited by
-
B-cell clonogenic activity of HIV-1 p17 variants is driven by PAR1-mediated EGF transactivation
Cancer Gene Therapy (2021)
-
Heparin and heparan sulfate proteoglycans promote HIV-1 p17 matrix protein oligomerization: computational, biochemical and biological implications
Scientific Reports (2019)
-
p17 from HIV induces brain endothelial cell angiogenesis through EGFR-1-mediated cell signalling activation
Laboratory Investigation (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
