
Abstract
Honeybee (Apis mellifera L.) workers act as passive vectors of Paenibacillus larvae spores, which cause the quarantine disease American foulbrood (AFB). We assessed the relative proportions of P. larvae within the honeybee microbiome using metabarcoding analysis of the 16 S rRNA gene. The microbiome was analyzed in workers outside of the AFB zone (control - AFB0), in workers from asymptomatic colonies in an AFB apiary (AFB1), and in workers from colonies exhibiting clinical AFB symptoms (AFB2). The microbiome was processed for the entire community and for a cut-off microbiome comprising pathogenic/environmental bacteria following the removal of core bacterial sequences; varroosis levels were considered in the statistical analysis. No correlation was observed between AFB status and varroosis level, but AFB influenced the worker bee bacterial community, primarily the pathogenic/environmental bacteria. There was no significant difference in the relative abundance of P. larvae between the AFB1 and AFB0 colonies, but we did observe a 9-fold increase in P. larvae abundance in AFB2 relative to the abundance in AFB1. The relative sequence numbers of Citrobacter freundii and Hafnia alvei were higher in AFB2 and AFB1 than in AFB0, whereas Enterococcus faecalis, Klebsiella oxytoca, Spiroplasma melliferum and Morganella morganii were more abundant in AFB0 and AFB1 than in AFB2.
Similar content being viewed by others

Introduction
American foulbrood (AFB) is a quarantine disease of the larvae and pupae of the honeybee, Apis mellifera L., and it is listed in the Terrestrial Animal Health Code by the Office International des Epizooties (OIE) of the World Organization for Animal Health1. The disease is caused by the gram-positive, spore-forming facultative anaerobic bacterium Paenibacillus larvae 2, originally described as Bacillus larvae 3, and it causes substantial economic losses to beekeepers4. The spores of P. larvae are extremely infectious, but colonies differ in their resistance to AFB outbreaks5. Honeybee workers are suitable for the early detection of AFB6,7,8. In a study investigating three honeybee pathogens in Spain, the detected P. larvae (as well as Melissococcus plutonius) prevalence was two-fold greater in adult bees than in brood samples8. Similar P. larvae spore loads are found in honeybees isolated from different parts of the colony6, 7, indicating homogeneity across different stages of the adult bee life cycle. However, Gillard et al.7 noted that bees collected at the hive entrance are of limited value for AFB diagnostics7. Thus, greater differences in spore loads occur at the level of individual workers9, and there is a correlation between the proportion of clinically diseased cells and number of infected workers9. Additional knowledge regarding P. larvae transmission methods and occurrence in honeybee colonies is required to better understand AFB, which notably contributes to the loss of colonies.
In recent years, numerous techniques have been developed to detect the economically important pathogen P. larvae. As noted in the review by de Graaf et al.10, these microbiological and molecular methods are applicable to brood samples, food reserves (honey, pollen and royal jelly), adult workers, and wax debris (de Graaf et al. 2010). The spores of P. larvae have been recovered from beeswax11 and detected in commercial pollen12. However, the list of reviewed techniques did not include high-throughput sequencing (HTS), which has become important for the study of honeybee and bumblebee microbiomes13,14,15,16. HTS has also been utilized to study interactions between parasites and honeybee-associated bacteria17, 18. Currently, HTS is being used to investigate the bacterial community of honeybees in relation to European foulbrood19.
The microbiome of mature honeybee workers roughly consists of approximately 109 bacterial cells13, and the composition of the bacterial community is likely influenced by the pathogen load in a colony. In the case of P. larvae, even a tolerant honeybee colony typically does not exceed 2.5 × 105 spores per g of honey5. If such a concentration of spores is present in hive food stores, sequencing analysis of the adults feeding on the contaminated honey or using it to feed the brood will be successful, and moreover, the total microbiome will be influenced by the number of pathogen sequences. In this study, HTS was employed to compare the relative proportions of P. larvae in the microbiomes of honeybee workers obtained from colonies with and without clinical symptoms as well as control samples from outside of the AFB zone. Varroa destructor infestation was also investigated as a factor in our statistical analyses. Additionally, we performed an AFB microbiome analysis of pupae. The presence of pathogenic P. larvae detected in commonly investigated sample types is demonstrated from a novel perspective in this study and the impact of P. larvae on the whole microbial community structure is also assessed.
Results
Overall, the identified bacterial taxa formed 116 and 114 operational taxonomic units (OTUs) at 97% similarity in the worker (Table S1) and pupae (Table S2) samples, respectively. After omitting the pupae samples, the dataset for the total worker honeybee microbiome contained 1,222,779 sequences, ranging from 14,741 to 68,869 sequences per sample. The dataset of the environmental and parasitic bacteria (cut-off microbiome) comprised 78,387 sequences, which formed 92 OTUs, and the sequence numbers ranged from 104 to 16,381 per sample.
Differences in the worker honeybee microbiome by AFB category
Krona projections (Fig. 1a–c) show differences in the cut-off microbiome structure among AFB0, AFB1 and AFB2; according to these illustrations, P. larvae comprised 3% in AFB0, 5% in AFB1 and 50% in AFB2. Although the number of P. larvae sequences differed little between AFB0 and AFB1 (Fig. 1d), the Krona projections indicate substantial differences in the cut-off microbiome structure. Figure 1d shows the number of P. larvae sequences in AFB0, AFB1 and AFB2; however, the statistical comparison was performed using the relative abundance of P. larvae (see OTU3 in Table 1).

Krona projections of the microbiome of honeybee workers based on analyses of the parasitic/environmental portion of the microbiome and comparison of P. larvae sequence numbers. Paenibacillus larvae is shown in red. Legend: (a) AFB0: control bees from outside the AFB zone with no signs of AFB, (b) AFB1: bees from asymptomatic colonies in AFB apiaries, (c) AFB2: bees from colonies exhibiting clinical symptoms of AFB, (d) comparison of the number of P. larvae sequences.
As a factor, AFB was found to have no or only a marginal effect on the number of OTUs in the total parasitic/environmental microbiome datasets; the inverse Simpson diversity index was not affected (Table 2). Redundancy (RDA) analyses, particularly the variance inflation factors (VIFs; Table S3), which sometimes exceeded 1,000, suggested a strong multicollinearity among the environmental variables. Upon further investigation, this multicollinearity was attributable to geographic coordinates, dominated by longitude. Varroosis levels could be added to the models alongside AFB levels. Therefore, we employed partial db-RDA models in which only the coordinates and bee sample collection times were conditioned. When we evaluated all of the OTUs and the pathogenic OTUs separately, varroosis was only important for the former dataset at P < 0.001. Both RDA models were significant (F = 2.82, P < 0.001 and F = 2.36, P < 0.001), which was primarily attributable to the effect of AFB. Interestingly, unlike the case for all OTUs (for which the first two axes were significant; P < 0.01), only the first RDA axis was significant for pathogenic OTUs (P < 0.001). In particular, the triplot visualization for all OTUs suggested a positive correlation between P. larvae (OTU3) and the colonies exhibiting clinical signs of AFB (AFB2) (Fig. 2), whereas the core bacteria Bifidobacterium asteroides (OTU8) and Lactobacillus mellis (OTU9), were negatively correlated with P. larvae (OTU3). Additionally, level 2 varroosis (varr2) was closely associated with AFB1 (Fig. 2). Upon constraining the analysis to pathogenic OTUs, the triplot pattern changed, although P. larvae (OTU3) remained correlated with AFB2. As shown in Fig. 3, the direction of the varroosis level 2 vector (varr2) differed from that of the AFB1 vector. The OTUs that were positively correlated with varroosis level were Spiroplasma melliferum (OTU16) and Morganella morganii (OTU18), whereas Klebsiella pneumoniae (OTU49) was correlated with AFB1. As a factor, AFB did not influence the distribution of bacteria in the worker microbiome (total microbiome) according to the analysis of molecular variance (AMOVA; Fs = 1.291, P = 0.195), and the sample variability was not significantly different based on the homogeneity of molecular variance test (HOMOVA; Bv = 0.460, P = 0.093). In contrast, the dataset for environmental and parasitic bacteria was significantly influenced by AFB according to the AMOVA (Fs = 4.466, P < 0.001). The Bonferroni-corrected values revealed significant differences (P ≤ 0.05) among all AFB-derived samples. There were no differences in sample variability according to the HOMOVA test (Bv = 0.569, P = 0.311).

Triplot visualization of principal coordinates in the RDA of the Apis mellifera worker microbiome. The analysis was based on the total microbiome dataset and included varroosis and AFB environmental factors. A correlation triplot containing sample scores given by the weighted sums of OTUs was constructed. The first two axes explained 25% of the total variability in the dataset as indicated by the bracketed percentage.

Triplot visualization of principal coordinates in the RDA of the A. mellifera worker microbiome. The analysis was based on the parasitic/environmental microbiome dataset and included varroosis and AFB environmental factors. A correlation triplot containing sample scores given by the weighted sums of OTUs was constructed. The first two axes explained 22% of the total variability in the dataset as indicated by the bracketed percentage.
METASTATS analyses confirmed the significant influence of AFB on the relative number of P. larvae (see OTU3 in Table 1) in the pathogenic/environmental dataset. Higher relative abundances of P. larvae were found in AFB2 colonies than in AFB1 and AFB0 colonies; there was no difference between AFB1 and AFB0. However, the AFB1 colonies did not differ significantly from control apiaries outside the zone (AFB0) in the relative number of P. larvae (see OTU3 in Table 1). The bacterial taxa with significantly higher relative abundances in AFB2 and AFB1 than in AFB0 were Citrobacter freundii (OTU28) and Hafnia alvei (OTU40). Enterococcus faecalis (OTU11), Klebsiella oxytoca (OTU128), Morganella morganii (OTU18) and Spiroplasma melliferum (OTU16) had higher relative abundances in AFB0 and AFB1 than in AFB2. Finally, K. pneumoniae (OTU49) and Melissococcus plutonius (OTU52) were present in greater numbers in AFB1 than in AFB0 and AFB2.
Honeybee pupae
The taxonomic compositions of pupae were visualized in the KRONA projections (see Figure S1), which show the proportions of bacteria in the investigated samples. The bacterial community of the pupae obtained from AFB2 was primarily formed by one eudominant OTU (Table S2). Paenibacillus larvae (OTU3) was present in the pupae of AFB2, whereas the following taxa were detected in AFB1 pupae: core bacteria Lactobacillus apis (OTU2), Gilliamella apicola (OTU5), Snodgrasella alvi (OTU7), and Frischella perrara (OTU15). Enterococcus faecalis (OTU11) was prevalent in the sampled pupae from the control (AFB0) honeybee colonies.
Discussion
In this study, we analyzed the occurrence of P. larvae in AFB-affected apiaries, including colonies exhibiting clinical signs, and in asymptomatic colonies, by employing a HTS approach and comparing the obtained data with those from control samples. Our samples consisted of worker honeybees, which are suitable for AFB diagnostics6, 20, 21 and facilitate both horizontal and vertical bacterial transmission22. Based on our results, in addition to commonly used microbiological techniques and molecular methods, such as PCR, qPCR and potential proteomics approaches10, HTS is useful for detection of P. larvae in honeybee samples. The great advantage of HTS is its ability to identify not only the bacterial pathogen P. larvae but also other bacteria, including both symbiotic and pathogenic/environmental bacteria, enabling us to demonstrate that AFB can influence the honeybee worker microbiome composition. The results of this study provide insight into the transmission of P. larvae and AFB disease development at the microbiome level. Due to the presence of P. larvae in control samples, we believe that P. larvae survives in honeybee colonies in an enzootic state23. Additionally, we visualized differences in the microbiome of pupae in terms of AFB occurrence in colonies using KRONA projections.
Explanation of P. larvae quantitation
The number of spores per adult honeybee depends on the presence or absence of clinical symptoms21. According to Gende et al.21, approximately 3,000 P. larvae spores per adult bee may represent the threshold for the appearance of clinical AFB symptoms, and the same authors observed at least a 2-fold reduction in the number of spores in bees from colonies without clinical symptoms relative to the number in bees from colonies with AFB symptoms21. Our investigation of the worker honeybee microbiome using HTS revealed approximately 9-fold greater numbers of P. larvae in workers from colonies with clinical symptoms compared with the numbers in workers from colonies that were asymptomatic but located at an AFB-diseased site (OTU3 in Table 1). In contrast, there was only a slight, statistically non-significant increase in P. larvae abundance in asymptomatic colonies compared with the abundance in colonies used as controls outside the protective zone. Differences in the proportion of P. larvae within the microbiome correlated well with KRONA projections depicting the microbiome composition after the removal of symbiotic bacteria (Fig. 1a–c). In summary, following the extraction of parasitic/environmental bacteria, the worker bee microbiome was notably influenced by P. larvae in bee colonies that exhibited clinical signs of AFB. These results are important for the interpretation and presentation of HTS data for pathogens of relatively low abundance, and this situation is specific to cases in which the pathogenic agent does not cause disease and is only transmitted by the host. Specifically, worker bees act only as vectors because P. larvae do not germinate in the adult gut24. Honeybee workers are known to drift between colonies, but they also rob weak and collapsing colonies25, 26. Robbing honey contaminated with P. larvae spores is an important factor in AFB transmission; larvae in the thief colony can be infected by feeding on stolen honey5, 27. According to Gillard et al.7, who utilized cultivation techniques, spores were detectable in relatively high numbers (greater than 25%) in asymptomatic colonies located in AFB apiaries7. Because some spores remain active in the honeybee gut for more than 2 months, honeybees are able to spread bacteria to other members of a colony over a long period of time, allowing increased infection to occur5. Thus, even a slight increase in the number of spores in a colony, which we observed for AFB-asymptomatic colonies, is critical for AFB development. However, AFB development is related to pathogen resistance levels in a particular colony; thus, in some colonies, more spores are needed to initiate the clinical signs of AFB5.
The presence of P. larvae in control samples indicates an enzootic state
One of our important findings is the presence of low numbers of sequences in control samples obtained outside the AFB zone. This result is supported by HTS analyses in other experiments (unpublished data) unrelated to AFB; in some colonies, we identified reads corresponding to P. larvae, whereas in others, no reads corresponding to P. larvae were identified. The enzootic occurrence of P. larvae has previously been suggested23. The recent analysis of a large spectrum of molecular markers suggested that certain endemic populations of P. larvae may adapt to the local honeybee population28. In this context, there is risk for AFB development due to the exchange of genetic honeybee material among colonies of unknown origin. According to Hansen and Rasmussen29, 11% of honey is contaminated with P. larvae, and importantly, of the honey samples investigated in their study, 9% were obtained from colonies lacking any signs of AFB symptoms in the same year or the following year29. A study examining the prevalence of three pathogens in Spanish apiaries using multiplex PCR detected P. larvae in 1.5 to 4.2% of transverse study (samples differing between seasons and over 2 years)8. These results reveal the spatial enzootic occurrence of the pathogen, which can occasionally overcome the resilience of colonies and result in outbreaks. Apicultural practices are key for controlling the spread of pathogens such as P. larvae, with consideration for possible swarming22. An experienced beekeeper is able recognize honeybee colonies that are threatened and prevent future damage stemming from the weakened colony, which is more susceptible to disease, including infection by P. larvae. Moreover, an experienced beekeeper can prevent swarming, which represents a risk for uncontrolled pathogen spread. Swarms of unknown origin are particularly dangerous in areas with high concentrations of honeybee colonies.
The influence of P. larvae on other bacteria and the lack of an association between AFB status and Varroa occurrence
Paenibacillus larvae is suggested to be inactive in honeybee workers24 and is therefore not expected to have any direct effects on the bacterial community in the gut. The results of the AMOVA of the entire dataset are consistent with this expectation. However, we observed some effects on the microbiome when we performed RDA analysis (Fig. 2). Although we included varroosis as a factor in our analyses, the presence of Varroa in colonies did not have an additive effect on the microbiome in combination with P. larvae. This result corresponds to the experimental results of Alippi et al.30, who found that Varroa is not a vector for P. larvae 30. In bumblebees, defense against different pathogenic agents involves the participation of the bacterial community, which varies with infection14,15,16. Moreover, the honeybee microbiome changes under the pressure exerted by harmful parasites17, 18. Therefore, the apparent lack of a connection between Varroa occurrence and AFB in the present study was unexpected.
Interestingly, we identified C. freundii and H. alvei as synergic bacterial taxa to P. larvae in our study, whereas E. faecalis, K. oxytoca, S. melliferum and M. morganii were antagonistic within the community of pathogenic/environmental bacteria (Table 1). These findings raise the question of whether C. freundii and H. alvei are secondary bacterial invaders associated with AFB. Although little information is available regarding these bacteria, some authors have suggested that C. freundii and H. alvei are pathogenic to honeybees31. Hafnia alvei has also been sporadically described in the honeybee gut and is suggested to be an opportunistic pathogen that potentially interacts with other bacteria32. The correlations observed with P. larvae based on RDA analysis suggest that these bacteria occur in colonies that are weakened by AFB disease. The fact that K. pneumoniae and M. plutonius were more abundant in workers from asymptomatic colonies than in either control bees or colonies with AFB symptoms implies an association of these bacteria with the initial stage of AFB development in the colony. The decrease in E. faecalis, K. oxytoca, S. melliferum and M. morganii populations with the increase in P. larvae suggests possible negative influences of the pathogen on these bacteria. The negative correlations of the core bacteria B. asteroides and L. mellis with P. larvae (OTUs 8 and 9 in Fig. 2) similarly suggest negative influences of the pathogen. Because many active mechanisms by which bacteria influence other microbes have been described33, studies on these interactions remain a challenge for future work. It is possible that some metabolites of P. larvae influence the abundance of different bacteria in the honeybee colony. Paenibacillus larvae has been shown to produce siderophores34, which represents an important example of a competitive mechanism that involves a cooperative behavior33.
Finally, our results demonstrate the influence of AFB on the composition of the honeybee pupa microbiome. Our data obtained from pupae are preliminary, and more detailed examinations are warranted. The results suggest the prevalence of L. apis, G. apicola, S. alvi, and F. perrara in asymptomatic colonies in AFB-diseased apiaries, whereas in the asymptomatic, control colonies only E. faecalis was present. In the future, it will be of interest to study the underlying mechanism of this influence to explain these differences in bacterial presence and determine whether these findings are generally valid. It is not surprising that colonies with clinical signs of AFB were dominated by P. larvae, and in cases where pupae survive to adulthood, these individuals should be considered vectors of P. larvae in their colony.
Methodological note – the cut-off microbiome
In this study, within the total microbiome, NGS was not an appropriate method to evaluate P. larvae in honeybee workers, but it was applicable for the analysis of the cut-off microbiome after the dominant core bacteria were reduced. This is because the sequences of symbiotic bacteria overlap with “rare” sequences of P. larvae. To enable data evaluation, we eliminated the core symbiotic bacteria from the microbiome; i.e., the sequences of core symbiotic bacteria were omitted from further analyses15, 16. Then, the pathogenic bacterium P. larvae was found to be prevalent in the bacterial community consisting of environmental/parasitic bacteria (Fig. 1). According to RDA analysis, P. larvae OTUs within the cut-off microbiome together with the presence of varroosis explained much of the variability in the bacterial microbiome and changes in the relative proportion of P. larvae, which were identifiable in both datasets (the total bacterial community and the community of pathogenic/environmental bacteria) (Figs 2 and 3).
Materials and Methods
Apiaries and sampling
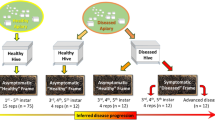
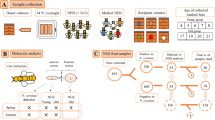
Apiaries were selected according to their presence in the AFB zones denoted by the State Veterinary Administration of the Czech Republic; note that an AFB zone in Czechia is defined as a 5-km flight radius surrounding a diseased apiary. For the samples, we coded AFB as follows: (i) AFB0 (control): bees from outside the AFB zone with no signs of AFB; (ii) AFB1: bees from asymptomatic colonies within AFB apiaries; and (iii) AFB2: bees from colonies exhibiting clinical AFB symptoms. See Table 3 for a description of colonies, apiaries, sampling dates, AFB infestations and varroosis levels. After sampling, colonies exhibiting AFB symptoms in AFB apiaries were burned according to the regulations of Czechia; therefore, subsequent sample collection was impossible. Samples of the European honeybee, A. mellifera carnica, were collected from brood combs into polypropylene bags. Samples from each colony comprised three biological replicates of 10 worker bees, and we also sampled 10 pupae (purple eyes) from each colony where available. Varroa infestation levels were described according to Hubert et al.18 based on mites falling onto the bottom boards of a colony following treatment. The colonies were classified into three categories of varroosis infestation: level 1, 0–50 mites (low Varroa infestation; VDI1); level 2, 51–100 mites (moderate Varroa infestation; VDI2), and level 3, 101 or more mites (high Varroa infestation; VDI3).
DNA extraction from honeybees
Each worker sample was surface-sterilized by washing in pure ethanol, followed by rinsing three times with sterile phosphate-buffered saline (3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, and 135 mM NaCl) containing 0.05% w/w Tween® 20 detergent (PBS-T) (Sigma-Aldrich, Saint Louis, MO, USA) to remove the surface microflora. The pupae were not surface-treated. Worker or pupal samples were homogenized (whole-body homogenates) in 6 mL of sterile PBS-T in a glass Potter-Elvehjem homogenizer (Kavalier, Sazava, Czechia). The homogenates were transferred to sterile tubes (cat no. D1003; KRD, Prague, Czechia) and centrifuged (CL31R, Thermo Fisher Scientific, Waltham, MA, USA) at 845 × g for 5 minutes. The supernatants were mixed with 6 mL of phenol/chloroform/isopropanol (Roti-Phenol®, cat no. A156.2, Carl Roth, Karlsruhe, Germany) and centrifuged at 3,381 × g for 5 minutes. This step was repeated with the upper aqueous phase, which was extracted twice with chloroform:isopropanol (24:1) and centrifuged. The upper aqueous phase was then transferred into Eppendorf tubes and precipitated with a 0.1 volume of 3 M sodium acetate (cat no. S7899, Sigma-Aldrich, Saint Louis, MO, USA) and a 0.74 volume of isopropanol. For precipitation, the mixture was incubated at −20 °C for 20 minutes. Then, the tubes were centrifuged at 18,242 × g for 15 minutes, and the pellets were washed twice in 70% pure ethanol. The dried pellets were re-suspended in 200 µL of ddH2O (56 °C) via pipetting. Then, the DNA was cleaned using a Geneclean® Turbo kit (cat no. 1102–600, MP Biomedicals, Santa Ana, CA, USA). The DNA samples were stored at −40 °C until use.
High-throughput sequencing (HTS)
The quality and presence of bacterial DNA in each sample was tested by performing PCR amplification using eubacterial primers, AFB primers and routinely used protocols35, 36. When amplicons were not obtained, the samples were replaced with new samples that were positive for amplicons. The DNA samples were sent to MR DNA for sequencing (http://mrdnalab.com, Shallowater, TX, USA). Sequencing of the V1–V3 portion of the 16 S rRNA gene was based on the 27Fmod and 519Rmod universal eubacterial primers on the Illumina MiSeq platform and the bTEFAP® process37. Read lengths were 300 bp, and forward and reverse reads were obtained. Sequences were processed as previously described18 using MOTHUR v.1.36.1 software38 according to the MiSeq standard operating procedure (MiSeq SOP39; http://www.mothur.org/wiki/MiSeq_SOP; accession date - 3/10/2016) and UPARSE with a USERCH pipeline40. The singletons and putative chimeras were discarded, and the OTUs were identified at 97% similarity according to the Ribosomal Database Project (http://rdp.cme.msu.edu) using training set no. 1541. Then, the representative sequences were processed using the BLASTn program on the NCBI platform (https://blast.ncbi.nlm.nih.gov/)42, and OTUs similar to chloroplasts and Archaea were removed. The best search hits were chosen based on the highest bit scores. Abundance data were then reincorporated into the dataset by mapping the initial sequences against the representative OTUs. The data were deposited in GenBank as SRA project no. SRP093442, and a list of samples is provided in Table S4. The taxonomic features of the samples were visualized by performing KRONA projections43, and abundance data were transformed into a shared file and processed in MOTHUR.
Data analyses
Based on the NCBI and RDP matches, we identified honeybee symbionts and pathogenic/environmental bacteria44 and constructed a shared file for all OTUs and a separate shared file for pathogenic/environmental OTUs according to the analyses of Cariveau et al.15, 16. The OTU classifications are presented in Tables S1 and S2. Both shared files were standardized by subsampling for a minimal number of sequences in sample, i.e., 14,741 sequences for all bacteria and 104 sequences for pathogenic/environmental bacteria in MOTHUR.
Alpha diversity was assessed by calculating the inverse Simpson index and the number of OTUs, and the effects of the factor AFB status were tested by performing the nonparametric Kruskal–Wallis test using XLSTAT software (http://www.xlstat.com/en/, Addinsoft, New York, NY, USA).
Beta diversity was studied by performing distance-based RDA (db-RDA), sometimes called the constrained analysis of principal coordinates. A Bray-Curtis dissimilarity matrix was used as the basis for this examination45. The main purpose was to determine if the factor of AFB zone substantially influenced only the pathogenic OTUs or all of the OTUs. Therefore, as with the AMOVA and HOMOVA analyses, two distinct OTU subsamples were used separately. In addition to the effects of AFB status, the effects of Varroa infestation level (factor with three levels) (Table 3) were explored. During the construction of db-RDA models, covariates such as geographic coordinates (longitude and latitude) and the time of bee sample collection were included, and VIF values46 were controlled for when adding environmental explanatory variables to the models. Moreover, the decision to include variables in the models was supported by the forward selection implemented in the “packfor” R package47 or permutational ANOVA-like tests performed with the same statistical package, the “vegan” R package48, with which the db-RDA models themselves were constructed. The partial db-RDA models were of particular interest in that they suppressed the potential risk of multicollinearity when regressing the response variables on the matrix composed of environmental variables. Additionally, the significance of canonical axes, the result of the PCA stage of the RDA, was determined by means of the permutational tests regardless of the proportion of variability that they explained. Regardless of their significance, the first two axes formed the basis of the correlation triplots in which site (sample) scores were expressed as the weighted sums of species given the dissimilarity matrix. Population-level analyses were carried out by comparing AFB zones and microbiomes for both the total bacteria and pathogenic/environmental bacteria datasets with METASTATS49 using 100,000 permutations in MOTHUR. To allow the results to be visualized more clearly, we have multiplied the vectors of the environmental variables AFB and varr2 2-fold and the OTU vectors 4-fold in the triplot visualizations presented in Figs 2 and 3.
References
OIE. Chapter 9.2. Infection of honey bees with Paenibacillus larvae (American foulbrood). In: OIE Terrestrial Animal Health Code, vol. 2 (OIE - World Organisation for Animal Health) http://www.oie.int/index.php?id=169&L=0&htmfile=chapitre_paenibacillus_larvae.htm (2016).
Ash, C., Priest, F. G. & Collins, M. D. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test: proposal for the creation of a new genus Paenibacillus. Antonie Van Leeuwenhoek 64, 253–260 (1993/1994).
White, G. F. The bacteria of the apiary, with special reference to bee diseases. (Bureau of Entomology, U. S. Department of Agriculture, 1906).
Genersch, E. American foulbrood in honeybees and its causative agent. Paenibacillus larvae. J. Invertebr. Pathol. 103, S10–S19 (2010).
Hansen, H. & Brodsgaard, C. J. Foulbrood diseases. Apiacta 34, 69–83 (1999).
Lindstrom, A. & Fries, I. Sampling of adult bees for detection of American foulbrood (Paenibacillus larvae subsp. larvae) spores in honey bee (Apis mellifera) colonies. J. Apic. Res 44, 82–86 (2005).
Gillard, M., Charriere, J. D. & Belloy, L. Distribution of Paenibacillus larvae spores inside honey bee colonies and its relevance for diagnosis. J. Invertebr. Pathol. 99, 92–95 (2008).
Garrido-Bailon, E. et al. The prevalence of the honeybee brood pathogens Ascosphaera apis, Paenibacillus larvae and Melissococcus plutonius in Spanish apiaries determined with a new multiplex PCR assay. Microb. Biotechnol 6, 731–739 (2013).
Lindstrom, A. Distribution of Paenibacillus larvae spores among adult honey bees (Apis mellifera) and the relationship with clinical symptoms of American foulbrood. Microb. Ecol. 56, 253–259 (2008).
de Graaf, D. C. et al. Standard methods for American foulbrood research. J. Apic. Res 52, 52.1.11, doi:10.3896/IBRA.1.52.1.11 (2013).
Machova, M. Resistance of Bacillus larvae in beeswax. Apidologie 24, 25–31 (1993).
Gochnauer, T. A. & Corner, J. Detection and identification of Bacillus larvae in a commercial sample of bee-collected pollen. J. Apic. Res 13, 265–267 (1974).
Moran, N. A. Genomics of the honey bee microbiome. Curr. Opin. Insect Sci. 10, 22–28 (2015).
Koch, H. & Schmid-Hempel, P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. USA 108, 19288–19292 (2011).
Cariveau, D. P., Powell, J. E., Koch, H., Winfree, R. & Moran, N. A. Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus). ISME J. 8, 2369–2379 (2014).
Cariveau, D. P., Powell, J. E., Koch, H., Winfree, R. & Moran, N. A. Erratum to “Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus) [ISME J. 8, 2369–2379 (2014)]”. ISME J. 8, 2550–2551 (2014).
Hubert, J. et al. Changes in the bacteriome of honey bees associated with the parasite Varroa destructor, and pathogens Nosema and Lotmaria passim. Microb. Ecol. 73, 685–698 (2017).
Hubert, J. et al. Comparison of Varroa destructor and worker honeybee microbiota within hives indicates shared bacteria. Microb. Ecol. 72, 448–459 (2016).
Erban, T. et al. Bacterial community associated with honeybees Apis mellifera affected by European foulbrood. PeerJ (in review) (2017).
Lindstrom, A., Korpela, S. & Fries, I. The distribution of Paenibacillus larvae spores in adult bees and honey and larval mortality, following the addition of American foulbrood diseased brood or spore-contaminated honey in honey bee (Apis mellifera) colonies. J. Invertebr. Pathol. 99, 82–86 (2008).
Gende, L. et al. Searching for an American foulbrood early detection threshold by the determination of Paenibacillus larvae spore load in worker honey bees. Bull. Insectol 64, 229–233 (2011).
Fries, I., Lindstrom, A. & Korpela, S. Vertical transmission of American foulbrood (Paenibacillus larvae) in honey bees (Apis mellifera). Vet. Microbiol. 114, 269–274 (2006).
Peters, M., Kilwinski, J., Beringhoff, A., Reckling, D. & Genersch, E. American foulbrood of the honey bee: occurrence and distribution of different genotypes of Paenibacillus larvae in the administrative district of Arnsberg (North Rhine-Westphalia). J. Vet. Med. B 53, 100–104 (2006).
Riessberger-Galle, U., von der Ohe, W. & Crailsheim, K. Adult honeybee’s resistance against Paenibacillus larvae larvae, the causative agent of the American foulbrood. J. Invertebr. Pathol. 77, 231–236 (2001).
Mill, A. C. et al. Clustering, persistence and control of a pollinator brood disease: epidemiology of American foulbrood. Environ. Microbiol. 16, 3753–3763 (2014).
Pfeiffer, K. J. & Crailsheim, K. Drifting of honeybees. Insect. Soc 45, 151–167 (1998).
Shimanuki, H. & Knox, D. A. Bee health and international trade. Rev. sci. tech. 16, 172–176 (1997).
Morrissey, B. J. et al. Biogeography of Paenibacillus larvae, the causative agent of American foulbrood, using a new multilocus sequence typing scheme. Environ. Microbiol. 17, 1414–1424 (2015).
Hansen, H. & Rasmussen, B. The investigation of honey from bee colonies for Bacillus larvae. Dan. J. Plant Soil Sci. 90, 81–86 (1986).
Alippi, A. M., Albo, G. N., Marcangeli, J., Leniz, D. & Noriega, A. The mite Varroa jacobsoni does not transmit American foulbrood from infected to healthy colonies. Exp. Appl. Acarol. 19, 607–613 (1995).
Lyapunov, Y. E., Kuzyaev, R. Z., Khismatullin, R. G. & Bezgodova, O. A. Intestinal enterobacteria of the hibernating Apis mellifera mellifera L. bees. Microbiology 77, 373–379 (2008).
Tian, B. & Moran, N. A. Genome sequence of Hafnia alvei bta3_1, a bacterium with antimicrobial properties isolated from honey bee gut. Genome Announc 4, e00439–16, doi:10.1128/genomeA.00439-16 (2016).
Hibbing, M. E., Fuqua, C., Parsek, M. R. & Peterson, S. B. Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25 (2010).
Hertlein, G. et al. Production of the catechol type siderophore bacillibactin by the honey bee pathogen Paenibacillus larvae. PLoS One 9, e108272, doi:10.1371/journal.pone.0108272 (2014).
Lane, D. J. 16S/23S rRNA sequencing. In: eds. Stackebrandt, E. & Goodfellow, M. Nucleic acid techniques in bacterial systematics. (John Wiley and Sons, 1991), p. 115–175.
Dobbelaere, W., de Graaf, D. C. & Peeters, J. E. Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp. larvae) using a 16S rRNA gene based PCR. Apidologie 32, 363–370 (2001).
Chiodini, R. J. et al. Microbial population differentials between mucosal and submucosal intestinal tissues in advanced Crohn’s disease of the ileum. PLoS One 10, e0134382, doi:10.1371/journal.pone.0134382 (2015).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42, D633–D642 (2014).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12, 385, doi:10.1186/1471-2105-12-385 (2011).
Engel, P. et al. The bee microbiome: impact on bee health and model for evolution and ecology of host-microbe interactions. mBio 7, e02164–02115, doi:10.1128/mBio.02164-15 (2016).
Engel, P. et al. Standard methods for research on Apis mellifera gut symbionts. J. Apic. Res. 52, UNSP 52.4.07, doi:10.3896/IBRA.1.52.4.07 (2013).
Kutner, M. H., Nachtsheim, C. J., Neter, J. & Li, W. Applied linear statistical models, 5 th edn. (McGraw-Hill Irwin, 2005).
Dray, S., Legendre, P. & Blanchet, G. packfor: Forward selection with permutation (Canoco p. 46). R-Forge, The R Project for Statistical Computing http://R-Forge.R-project.org/projects/sedar/ (2013).
Oksanen, J. et al. vegan: Community Ecology Package. CRAN - The Comprehensive R Archive Network http://CRAN.R-project.org/package=vegan (2016).
White, J. R., Nagarajan, N. & Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5, e1000352, doi:10.1371/journal.pcbi.1000352 (2009).
Acknowledgements
The authors are grateful to the beekeepers and to the State Veterinary Administration of the Czech Republic (http://eagri.cz/public/web/en/svs/portal/) for allowing us to collect bee samples during the outbreak. We thank Marie Bostlova and Jan Hubert Jr. for their technical assistance. The study was supported by project no. QJ1310085 of the Ministry of Agriculture of the Czech Republic (http://www.eagri.cz). We thank the editors at American Journal Experts for English language editing.
Author information
Authors and Affiliations
Contributions
Wrote the main manuscript: T.E. and J.H. Conceived of and designed the experiments: J.H., M.K., E.T. and D.T. Collected samples: J.H., M.K., M.N. and J.T. Performed laboratory analyses and evaluated data: M.N., J.H. and B.H. Performed statistical analysis: O.L. and J.H. Prepared the figures and tables: J.H., O.L., E.T., M.M. and B.H. Edited the body of the manuscript and references: M.M. Provided grants and materials: T.E. and D.T. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Erban, T., Ledvinka, O., Kamler, M. et al. Honeybee (Apis mellifera)-associated bacterial community affected by American foulbrood: detection of Paenibacillus larvae via microbiome analysis. Sci Rep 7, 5084 (2017). https://doi.org/10.1038/s41598-017-05076-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05076-8
This article is cited by
-
Probiotic candidates for controlling Paenibacillus larvae, a causative agent of American foulbrood disease in honey bee
BMC Microbiology (2023)
-
Alterations in the Microbiota of Caged Honeybees in the Presence of Nosema ceranae Infection and Related Changes in Functionality
Microbial Ecology (2023)
-
Hexanic extract of Achyrocline satureioides: antimicrobial activity and in vitro inhibitory effect on mechanisms related to the pathogenicity of Paenibacillus larvae
Veterinary Research Communications (2023)
-
Double-coated microencapsulation of honeybee endogenous probiotics as a new strategy for the biocontrol of the American foulbrood disease
Egyptian Journal of Biological Pest Control (2022)
-
Establishment of apiary-level risk of American foulbrood through the detection of Paenibacillus larvae spores in pooled, extracted honey in Saskatchewan
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
