
Abstract
Clubroot, caused by Plasmodiophora brassicae, is an important disease of Brassica crops worldwide. F1 progeny from the Brassica rapa lines T19 (resistant) × ACDC (susceptible) were backcrossed with ACDC, then self-pollinated to produce BC1S1 lines, From genotyping-by-sequencing (GBS) of the parental lines and BC1 plants, about 1.32 M sequences from T19 were aligned into the reference genome of B. rapa with 0.4-fold coverage, and 1.77 M sequences with 0.5-fold coverage in ACDC. The number of aligned short reads per plant in the BC1 ranged from 0.07 to 1.41 M sequences with 0.1-fold coverage. A total of 1584 high quality SNP loci were obtained, distributed on 10 chromosomes. A single co-localized QTL, designated as Rcr4 on chromosome A03, conferred resistance to pathotypes 2, 3, 5, 6 and 8. The peak was at SNP locus A03_23710236, where LOD values were 30.3 to 38.8, with phenotypic variation explained (PVE) of 85–95%. Two QTLs for resistance to a novel P. brassicae pathotype 5x, designated Rcr8 on chromosome A02 and Rcr9 on A08, were detected with 15.0 LOD and 15.8 LOD, and PVE of 36% and 39%, respectively. Bulked segregant analysis was performed to examine TIR-NBS-LRR proteins in the regions harboring the QTL.
Similar content being viewed by others


Introduction
The soil-borne pathogen Plasmodiophora brassicae Woronin causes clubroot disease in Brassica oilseed and vegetable crops worldwide. It belongs to the Infra Kingdom Rhizaria, which is a diverse group of amoeboid protists1. The haploid resting spores of P. brassicae release zoospores that infect root hairs, in which multi-nucleate plasmodia are formed. These plasmodia develop into uninucleate secondary zoospores, which are released into the soil and then infect young roots. Secondary multi-nucleate plasmodia develop rapidly and colonize the root cortex2, which stimulates the host plants to produce the characteristic root galls, known as clubs. Disorganization of tissues in the clubbed roots restricts the flow of water and nutrients, resulting in above-ground symptoms that include stunting, yellowing, premature senescence, and reduction in both seed yield and quality3. The plasmodia become sessile and divide to produce resting spores, which are released from decaying clubs into the soil, where they can survive for many years2. The combination of prolonged survival in the absence of a wide host range including many weed species, and relative insensitivity to most reduced-risk fungicides and bactericides makes it difficult to manage clubroot using crop rotation or anti-microbial seed treatments4. Therefore, genetic resistance is generally considered to be the most effective approach for clubroot management.
Clubroot is a serious constraint to canola (Brassica napus L.) production on the Canadian prairies. In Canada, five pathotypes of P. brassicae (pathotypes 2, 3, 5, 6 and 8) have been identified based on the differential system of Williams5, with pathotype 3 the most prevalent on canola in the prairie region6,7,8. The first clubroot-resistant canola cultivar in western Canada was released in 2009, and was followed by the release of other resistant cultivars from various seed companies starting in 2010. These cultivars exhibited strong resistance to the pathotypes of P. brassicae present in Canada9. However, resistance in Canadian canola cultivars has been overcome recently by new strains of the pathogen identified in Alberta10. These new strains have been informally designated as pathotype 5x because they are classified as pathotype 5 on the differentials of Williams (1966)5 but (unlike the original pathotype 5) are highly virulent on clubroot resistant canola.
Lines with resistance to a broad range of pathotypes of P. brassicae have been identified in the canola progenitor species B. rapa 11, 12. This species could be used to broaden the genetic base of clubroot resistance in canola. Introgression of traits from B. rapa into canola through interspecific crosses is possible13,14,15,16,17, so resistance to clubroot from B. rapa could be transferred into canola through conventional breeding.
The identification and genetic mapping of clubroot resistance genes has been carried out in B. rapa 18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34, B.oleracea 35,36,37,38,39,40,41,42,43,44 and B. napus 45,46,47,48,49,50,51. Two resistance genes, CRa and Crr1, have been isolated from Chinese cabbage lines of B. rapa. They encode Toll-Interleukin-1 receptor/nucleotide-binding site/leucine-rich-repeat (TIR-NBS-LRR, TNL) proteins52, 53.
Genotyping-by-sequencing (GBS) offers a new tool to explore the genetic control of complex traits. GBS analysis was used in the current study to: 1) characterize genome-wide variants in B. rapa; 2) identify SNP sites that could be used for genetic mapping; and 3) detect QTL effectively resistant to multiple pathotypes of P. brassicae identified in western Canada; and 4) identify possible candidate genes for each QTL.
Results
Resistance to clubroot in B. rapa canola line T19 and BC1S1 progenies
The B. rapa canola line T19 was shown to be highly resistant to pathotype 3 while ACDC was highly susceptible (Fig. 1a). To determine if T19 was resistant to the other pathotypes present in Canada, we inoculated T19 plants with six Canadian pathotypes of P. brassicae. This line was found to be highly resistant to all of the pathotypes (0% DSI), while ACDC and B. rapa subsp. pekinensis (European Clubroot Differential (ECD) 05, a susceptible check) were highly susceptible (100% DSI) (Fig. 1b).

Evaluation of parental lines and BC1S1 population derived from ACDC × (ACDC × T19) for resistance to clubroot. (a) R and S phenotypes in the parental lines with pathotype 3; (b) Disease severity indexes (DSIs) of the parental line and susceptible control ECD 05 with pathotype 2, 3, 5, 6, 8 and P5x of Plasmodiophora brassicae; (c) Distribution of DSIs in the BC1S1 population consisting of 92 lines.
Ninety two BC1S1 lines derived from 92 BC1 plants were then tested for resistance to each of the pathotypes. All seedlings of the susceptible check (ECD 05) and the susceptible parent ACDC developed severe clubbing (100% DSI). The resistant parent, T19, was resistant to each pathotype (0% DSI, Supplementary Fig. S1), confirming that T19 was highly resistant. The distribution of clubroot severity in response to inoculation with pathotypes 2, 3, 5, 6 and 8 (Fig. 1c, Supplementary Fig. S1) could be divided into two major classes: resistant (R) lines with DSI < 60% and susceptible (S) lines with DSI > 60%. Of the 92 lines, 49 lines were classified as R and 42 lines as S to each of the pathotypes (Fig. 1c). The segregation of R and S fitted a 1:1 ratio (X2 = 0.39; P = 0.53), indicating that resistance to the pathotypes may be controlled by a single resistance gene in the R parent line T19. DSIs in the population were highly correlated (r ≥ 0.92) among the five pathotypes (Table 1), indicating that resistance to the pathotypes may be controlled by the same gene or closely linked genes. However, 71 of the 92 lines were classified as R and 21 lines as S to pathotype 5x. Segregation for R and S in the population did not fit a 1:1 ratio (X2 = 27.2; P < 0.001), but fit a 3:1 ratio (X2 = 0.23; P = 0.63). This indicates that resistance to pathotype 5x may be controlled by two resistance genes in T19. A correlation of the DSIs in response to 5x with those to the rest of the pathotypes was not found (r ≤ 0.06) (Table 1), indicating that the genes for resistance to pathotype 5x are not linked to the gene for resistance to pathotypes 2, 3, 5, 6 and 8.
Alignment of DNA short reads into the B. rapa reference genome
Approximately 1.32 million (M) sequences were aligned into the chromosomes of the reference genome in the resistant parent T19 and 1.77 M sequences in the susceptible parent ACDC (Table 2). The accumulated length of sequences was 98.3 Mb with 0.4-fold coverage in T19, and 131.1 Mb with 0.5-fold coverage in ACDC. The average aligned short read length was 74.3 bases in T19 and 74.5 bases in ACDC (Table 2).
The number of aligned short reads from each sample in the BC1 population varied, ranging from 0.07 to 1.41 M sequences (Fig. 2). The accumulated length of sequences in the 92 samples was 3076.4 Mb with 12.0-fold coverage. However, the average accumulated length of sequences was 33.4 Mb with 0.1-fold coverage (Table 2).

Numbers of sequences aligned into the Brassica rapa reference genome and variants (SNPs and InDels) identified in each sample in comparing with the DNA sequence of the reference genome.
Identification of variants in the population
There was a strong positive correlation (r = 0.99) between the numbers of variants compared with the number of sequences aligned into the reference genome (Fig. 2), indicating that the number of variants identified each sample was associated with sequencing depth. However, the proportions of SNPs and InDels were similar in the parental lines and BC1 population, with about 88% and 12%, respectively (Table 2, Supplementary Fig. S2). Variants in both T19 and ACDC samples were frequent, with 126.3 K and 162.1 K respectively (Table 2). The numbers of variants in the BC1 population were in a range of 14 to 143 K (Fig. 2) with a mean of 55.3 K variants per sample (Table 2).
Identification of polymorphic SNP sites in the BC1 population and construction of linkage groups
There were 16,618 SNPs and 2,127 InDels identified in at least 46 of the 92 BC1 samples (50% of the samples). Since the susceptible parent, ACDC, is a doubled haploid line, all SNP sites should theoretically be homozygous. However, out of 16,618 SNP sites, 3,263 SNP sites (19.6%) had a heterozygous genotype. Monomorphic phenotypes between the parents or among the 92 individuals were identified in 8,392 SNP sites (50.5%) (Supplementary Table S1). The remaining 4963 SNP sites were further analyzed using software JoinMap 4.1. A total of 1584 SNP sites, accounting for 9.5% of 16,618 SNP sites, could be assigned into 10 chromosomes of B. rapa. A genetic map consisting of 1584 SNP sites distributed to 10 chromosomes of B. rapa was constructed (Fig. 3). The number of SNP sites was in a range of 120 to 219, with an average number of 157 on each chromosome in the map. The length of each chromosome ranged from 263.15 to 709.51 cM with an average length of 479.3 cM. The SNP interval of each chromosome ranged from 2.07 to 3.98 cM with an average of 3.05 cM (Supplementary Table S2).

The linkage map of B. rapa consisting of 1584 SNP sites.
Identification of QTL for resistance to six pathotypes of P. brassicae
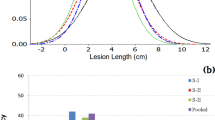
Mapping of the QTLs was performed using the linkage map (Fig. 3) and trait values for resistance to the six pathotypes of P. brassicae. QTL mapping analysis resulted in three QTLs being detected in chromosomes A02, A03 and A08 (Table 3). A single co-localized QTL, designed as Rcr4, was detected for resistance to pathotypes 2, 3, 5, 6 and 8, but not to pathotype 5x, which was located on chromosome A03 (Fig. 4). Rcr4 spanned from 267.48 to 310.75 cM with a peak position at 282.65 cM. The SNP site nearest the peak for Rcr4 was at SNP site A03_23710236 (chromosome_physical location) with LOD values of 30.3 to 38.8 for the five pathotypes, and phenotypic variation explained (PVE) 85% to 94% (Table 3). Two QTLs, designated Rcr8 and Rcr9, were detected on chromosomes A02 and A08, respectively, for resistance to pathotype 5x. Rcr8 spanned from 126.91 to 164.4 cM, with a peak position at 139.8 cM. The SNP site nearest the peak for Rcr8 was at SNP site A02_18552018 (15 LOD), with PVE of 36%. Rcr9 spanned from 214.61 to 291.25 cM, with a peak position at 242.5 cM. The SNP site nearest the peak for Rcr9 was at A08_10272562 (15.8 LOD) with PVE of 39% (Table 3). As show in Table 3, the values of additive for the three QTL were positive, indicating that the resistant loci were derived from the resistant parent T19.

Three significant QTL of Rcr4, Rcr7 and Rcr8 detected chromosomes on A03, A02 and A08, respectively.
Identification of TNL genes and variants in the genes in the target regions
DNA short reads from selected BC1 individuals, based on clubroot severity and the presence of SNP alleles in the respective identified QTL peaks, were pooled and aligned into the reference genome (Supplementary Table S3). The TNL genes in the target regions were identified (Table 4). The numbers of poly variants in the TNL genes of the Rcr4, Rcr8 and Rcr9 intervals were assessed because the poly variants represent differences in the DNA sequences between the R and S bulks. SeqMan Pro software was used to sort variants that affected amino acid sequences into four groups: non-synonymous, nonsense, frameshift and synonymous variants. However, nonsense and frameshift variants were not found in any of the TNL genes in this study.
Rcr4 was mapped into chromosome A03 in the genetic region of 267.48 to 310.75 cM (Table 3), corresponding to the physical region at A03_22692045 to A03_25649385 base, spanning about 2.96 Mb and including 441 genes based on the B. rapa reference genome. Six genes (Bra012541, Bra019413, Bra019412, Bra019410, Bra019409 and Bra019273) as shown in Table 4, encode TNL-class disease resistance proteins in this region. No poly variants were identified in Bra019412 and Bra019273. A total of 13 poly variants were identified from the coding sequences in the rest of four TNL genes (Supplementary Table S4). Three non-synonymous poly variants were identified in Bra019413 and one in Bra019410. Only synonymous SNPs were found in Bra012541 and Bra019409 (Table 4). These five genes locate on chromosome A03 from 23,717,282 from 25,352,019, in an interval of 1.63 Mb.
Rcr8 was mapped in the region of 126.91 to 164.4 cM (Table 3), corresponding to a physical region at A02_18503233 to A02_22097179 base, spanning about 3.59 Mb and including 396 genes. Four genes (Bra022069, Bra022071, Bra026556 and Bra032996) encode TNL-class disease resistance proteins in the interval (Table 4). However, no DNA variants were identified in the genes. These five genes are located on chromosome A02 from 18,690,084 from 22,085,693, in an interval of 3.40 Mb.
Rcr9 was mapped in the region of 214.61 to 291.25 cM (Table 3), corresponding to a physical region at A08_7105657 to A08_13587639 base, spanning about 6.48 Mb and including 838 genes. Only two genes (Bra020936 and Bra020861) are known to encode TNL-class disease resistance proteins in this region (Table 4). However, no poly variants were identified in the genes (Table 4). These two genes are located on chromosome A08 from 10,295,068 from 10,825,238, in an interval of 0.53 Mb.
Discussion
GBS generates a wealth of short DNA sequence reads from random places in the genome. The current study characterized variants in the B. rapa population, identified SNP sites that can be used for genetic mapping, and detected QTLs for resistance to specific clubroot pathotypes. SNP variants were the most common types of DNA sequence variation, accounting for 88% of the variation in the B. rapa genome. The number of variants identified in each sample was correlated strongly with sequencing depth. These observations are consistent with a previous study using RNA-seq33. Although coverage was low in the current study, variants from the reference B. rapa genome54 were very frequent, with a mean of 55 K variants per plant in the BC1 population. One factor that likely contributed to this high frequency of variants was that the parental lines were canola, while the reference genome was a Chinese cabbage cultivar.
The construction of genetic maps in the early 1990s based on DNA markers led to an explosion of activity directed toward the identification of QTLs in crop species. In addition to phenotyping individual lines, QTL mapping usually requires the identification of genome-wide polymorphic DNA markers for linkage analysis. However, the process for screening and genotyping polymorphic markers through analysis of markers is labour-intensive and time-consuming55. Progress in next generation sequencing (NGS) allows the identification of DNA markers through GBS. In the current study, the susceptible parent, ACDC, was a doubled haploid line, so each SNP site from this line should have been homozygous. However, numerous heterozygous SNP sites were identified. After filtering out the false heterozygous SNP and monomorphic SNP sites, some of the SNP sites could not be assigned into the chromosomes of B. rapa. Similarly, only small portion of GBS SNP sites that could be used for QTL mapping was also observed from the previous study44. The causes of heterozygous SNP sites in ACDC and the SNP sites that could not be assigned into any chromosomes by JoinMap 4.1 are likely errors from DNA sequencing or alignment of short reads. NGS can generate large amounts of data with problems such as high per-base error rates and non-uniform coverage, together with platform-specific read error profiles and artifacts56. In addition, triplication occurs in the B. rapa genome54, which could result in an ambiguous alignment that potentially leads to biases and errors in variant assessment. Identifying and correcting these issues imposes several statistical and computational challenges for the reliable detection of variants from NGS data56.
The identification of SNPs for bi-parental mapping of QTL through GBS has been carried out in several crops57,58,59 including B.oleracea 44. After filtering, 1,584 polymorphic SNP sites distributed to 10 chromosomes of B. rapa were identified. In the current study, 92 BC1 plants were assessed with a mean of only 0.1-fold coverage. Despite this low coverage, a set of high quality SNP sites and three strong QTLs were identified, indicating that identification of SNP markers through GBS is a cost effective and efficient method for QTL mapping.
The clubroot-resistant canola cultivars in western Canada were released in 2009 and 2010. These cultivars exhibited strong resistance to pathotypes 2, 3, 5, 6 and 8 of P. brassicae present in Canada. However, resistance in all of the Canadian canola cultivars has been overcome by new strains of the pathogen identified in Alberta after three years cultivation. The breakdown of resistance could be due to resistance in the cultivars controlled by a dominant gene originating from the same source therefore new sources of resistance is urgently needed. Identification of three QTL in this study facilitates breeders to develop canola cultivars carrying multiple resistance genes with more durable resistance to clubroot.
Several recent studies in Canada have focused on identifying sources of clubroot resistance and molecular markers to transfer clubroot resistance into canola18, 21, 33, 48,49,50. Although at least six pathotypes of P. brassicae occur in Canada, the research has been focused on resistance to pathotype 321, 48,49,50, which is the predominant pathotype on the Canadian prairies9. A clubroot resistance gene, Rcr1, with efficacy against pathotype 3, was mapped to chromosome A03 of B. rapa in the pak choy cultivar ‘Flower Nabana’21 and resistance to pathotypes 2, 5, 6 and 8 was associated with Rcr1 region on chromosome A0333. A resistance gene on chromosome A08 of rutabaga (B. napus subsp. napobrassica) also conferred resistance to these five pathotypes50. In the current study, resistance to the five pathotypes was highly correlated and likely controlled by one gene or tightly linked cluster of genes when assessed using conventional genetic analysis. One co-localized major QTL, Rcr4, was identified using GBS. This supported the hypothesis that a single gene or tightly linked genes controlled resistance to the five pathotypes. Since single spore isolates of pathotype 5x were not available, a P. brassicae population L-G02 orginating from a clubbed root was used in this study. P. brassicae is assumed to exist as mixtures of pathotypes in the soil, and even in single clubbed roots different pathotypes have been detected60. However, different pathotypes were not identified based on Williams’ differential and the Canadian resistant cultivars10. Resistance to the novel pathotype 5x was not correlated with resistance to the other five pathotypes. Genetic analysis indicated that resistance to pathotype 5x was likely controlled by two unlinked genes (Rcr8 and Rcr9) in the resistant line T19. These two loci for resistance to 5x were mapped onto chromosomes A02 and A08, respectively. This provides strong evidence that the two genes control resistance to pathotype 5x independently. Also, Rcr9 on A08 appears to differ from the resistance gene on A08 identified in rutabaga, because Rcr9 was effective against pathotype 5x but not against pathotypes 2, 3, 5, 6 or 8, while the resistance gene from rutabaga was effective against these five pathotypes50.
The effect of population size on QTL detection was conducted by Bradshaw et al.61. They evaluated floral morphology traits in monkey flower using populations of 93 and 465 F2 individuals. For QTLs common to the two populations, the estimate of effect size was reduced in the larger population. It is possible that the magnitude of QTL effects is overestimated in small populations. In this study, three strong QTL were identified by using the BC1 population consisting of 92 plants. Therefore, further studies on the effects of the Rcr4, Rcr8 and Rcr9 to clubroot resistance would be needed. A small F2 population with 78 plants was previously used for identifying QTL for resistance to clubroot in B. oleracea through GBS44.
Two cloned clubroot resistance genes, CRa and Crr1, have been reported previously to encode TNL proteins52, 53. Therefore, DNA changes in the TNL genes in the Rcr4, Rcr8 and Rcr9 target regions were examined. However, a precise determination of the candidate genes was not possible since very few variants were identified in the TNL genes. The reason for this is due to the low depth of sequencing in this study. In contrast, the candidate genes for Rcr1 were well predicted based on DNA variants among the TNL genes in the target region through deep sequencing33. As the cost for NGS is declining, deeper sequencing will become more affordable so higher depth sequencing would be recommended in the future research. Nonetheless, we could further narrow the gene intervals from 2.96 to 1.63 Mb for Rcr4, 3.59 to 3.4 Mb for Rcr8 and 6.48 to 0.53 Mb for Rcr9 by identifying the TNL genes in the respective QTL regions. Clubroot resistance gene Rcr1 was mapped to the same region as Rcr4, although Rcr1 was identified in pak choy (B. rapa subsp. chinensis)21, 33 while Rcr4 was identified from canola. Identifying the TNL gene(s) that correspond with Rcr4 or Rcr1, and the relationship between Rcr4 and Rcr1 will be addressed after the genes have been cloned. Four TNL genes were found in the Rcr8 region A02, spanning the interval of 18,503,233 to 22,097,179. In a previous study, CRc was mapped on A02 by QTL analysis and molecular marker m6R, which is located in the position of 2,112,653 to 2,113,153 of chromosome A02, and was shown to be closely linked to CRc 30. The difference in mapping location between Rcr8 and CRc demonstrates that these two resistance genes are not allelic. Two TNL genes, Bra020936 and Bra020861, were identified in the Rcr9 interval. The cloned clubroot resistance gene Crr1 is highly homologous to Bra020861 in the B. rapa reference genome53. Identifying the TNL gene(s) that corresponds with Rcr9 and the relationship between Rcr9 and Crr1 will also be addressed after the genes have been cloned.
Materials and Methods
Plant materials
T19, a B. rapa canola breeding line with clubroot resistance originating from the German turnip cultivar ‘Pluto’, was developed at the Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada (SRDC, AAFC), Saskatoon, Saskatchewan, Canada. T19 was crossed to ACDC, a clubroot-susceptible doubled-haploid, self-compatible B. rapa canola breeding line also developed at the SRDC. The resulting F1 plants were backcrossed with ACDC to produce BC1. Plants in the BC1 population were self-pollinated to produce BC1S1. To overcome self-incompatibility in B. rapa, bud-pollination was performed or each stigma was sprayed with 3% salt solution to produce sufficient seeds for evaluation of plant reaction to pathotypes 2, 3, 5, 6, 8 and 5x of P. brassicae. Ninety two BC1S1 lines were assessed in the study.
Isolates of P. brassicae and evaluation of BC1S1 population for resistance to clubroot
Similar method and experimental design as described by Suwabe et al.31 were used in this study. Twelve plants from each of BC1S1 lines, parental lines T19 and ACDC were tested for resistance to five single-spore isolates of P. brassicae, SACAN-ss3 (pathotype 2), SACAN-ss1 (pathotype 3), ORCA-ss4 (pathotype 5), AbotJE-ss1 (pathotype 6), and ORCA-ss2 (pathotype 8) under controlled conditions at the University of Alberta, Edmonton, Alberta, Canada. Resting spores of each isolate were extracted from the frozen galls as described10 and adjusted to a concentration of 5.0 × 107 resting spores/mL for each of the isolates. One-week-old seedlings of the host genotypes, which were pre-germinated on moistened filter paper in Petri dishes, were inoculated by dipping the entire root system in the resting spore suspension for 10 sec. The inoculated seedlings were then immediately planted in 6 cm × 6 cm × 6 cm plastic pots filled with Sunshine LA4 potting mixture, with one seedling per pot. Pots were thoroughly watered and transferred to a greenhouse at 21 °C ± 2 °C with a 16-h photoperiod. The potting mixture in the pots was kept saturated with water for the first week after inoculation and then watered and fertilized as required. Also, the reaction of each BC1S1 line and the parental lines to the P. brassicae population L-G0210, representing pathotype 5x, was tested on 14 to 20 plants per line under controlled conditions at SRDC, following the method described21. The experiments for pathotype 5x were repeated twice with similar results and only the first experiment was used for correlation study and identification of QTL. A Canadian cultivar “45H29” that can differentiate pathotypes 5 and 5x was included in the pathology experiments related to pathotype 5x.
Six weeks after inoculation, the roots of each line were dug out, washed with tap water, and examined for club formation. Clubroot severity was evaluated on a 0 to 3 scale as described previously62, where 0 = no clubbing, 1 = a few small clubs, 2 = moderate clubbing, and 3 = severe clubbing. A disease severity index (DSI) was calculated for each host line, using the method of Horiuchi and Hori63 as modified by Strelkov et al.6:
Correlation coefficients among the DSIs values in BC1S1 families to six pathotypes of P. brassicae were calculated using Microsoft Excel. The significance of the correlation coefficients was determined though t-tests64.
Based on our previous observation33, BC1S1 lines with DSI < 60% were likely from resistant BC1 plants. We therefore classified BC1S1 lines with DSI < 60% as R and those lines with DSI > 60% as S lines in this study.
DNA sequencing and alignment of reads to reference genome
DNA was extracted from young leaves of each of the 92 BC1 plants and parental lines following DNeasy Plant Mini Handbook (QIAGEN). The DNA samples from the 92 BC1 plants and 2 replications of the parental lines T19 and ACDC were sequenced by an Illumina HiSeq 2000 SE single-end lane at Data2Bio (Ames, IW, USA). The program SeqMan NGen 13 (DNASTAR, Madison, WI, USA) was used for short read assembly. Standard assembling and filtering parameters were used. Short reads from each of 92 BC1 samples and the combined two replicates of each parental line were aligned to the reference genome Brapa_sequence_v1.5.fa downloaded from: http://brassicadb.org/brad/downloadOverview.php. The reference genome consists of 10 chromosomes and 40,357 scaffolds. The total lengths of chromosomes and scaffolds are about 258 Mb and 27 Mb, equivalent to about 90% and 10% of the reference genome, respectively. To simplify data analysis, only the 258 Mb chromosome sequences were used in the current study.
Identification of variants, variant filtering, construction of linkage map and QTL mapping
Discovery of variants (SNPs and InDels) in comparison with the DNA sequences in the B. rapa ‘Chiifu’54 from each BC1 sample was performed using SeqMan Pro 13 (DNASTAR, Madison, WI, USA). Comparison of the variants among the 92 BC1 samples was carried out using Qseq 13 (DNASTAR, Madison, WI, USA). Only SNPs were used for further examination. Detected GBS-SNP sites were named based on the reference chromosome and position on the reference chromosome sequences. A SNP site was called in a given sample at following criteria: depth >5, Q > 10 and SNP percentage >10%. The remaining SNP sites after filtering were further analyzed using JoinMap 4.165. SNP alleles from the resistant parent T19 were scored as “h” and those from the susceptible parent ACDC as “a”. The Mendelian segregation distortion of each marker was examined using X2 test in JoinMap 4.1 and distorted markers were excluded from further analysis. Marker orders and positions in the genetic map were determined using maximum likelihood in Kosambi’s model with a minimum logarithm of odds (LOD) of ten. This set of SNP sites were used for interval mapping of QTLs for resistance to clubroot. A map was drawn using Mapchart 2.166 based on the genetic location determined by JoinMap 4.1. Mapping of resistance to the six pathotypes was performed using MapQTL 6 (www.biometis.wur.nl) with the interval mapping method. The LOD score threshold was initially set at 3.0 for QTL declaration, and QTLs that exceeded this LOD threshold were considered as suggestive QTLs. If any relevant QTL was identified, the LOD score threshold was determined using the 1,000-permutation test with a confidence of 0.99. QTL with LOD scores greater than the thresholds 4.14, 4.09, 4.15, 4.61, 4.03 and 4.03 (Table 3) for resistance to pathotypes 2, 3, 5, 6, 8 and 5x, respectively, at a confidence of 0.99, were declared significant. The QTL effects were estimated as PVE by the QTL.
Identification of DNA variants in the TNL genes in the target region
The poly variants that uniquely occurred in the R bulks but not in the S bulks with depth >5 in both samples were assessed further. Bulked segregant analysis was performed to identify DNA variants in the respective QTL intervals. Based on both phenotyping and genotyping results, selected samples of the 92 BC1 plants were classified as R or S. Short reads from R and S samples were pooled to form R and S bulks, and then each bulk read was aligned to the B. rapa reference genome. Identification of poly variants was carried out as described by Yu et al.33. Gene annotation was analyzed with Blast2GO67. The NBS-LRR genes described as Chalhoub et al.68 in the target region were also examined. Further confirmation of the genes with TNL domains was performed with Arabidopsis thaliana WU-BLAST2 Search at http://www.arabidopsis.org/wublast/index2.jsp.
References
Nikolaev, S. I. et al. The twilight of Heliozoa and rise of Rhizaria, an emerging supergroup of amoeboid eukaryotes. Proc. Natl. Acad. Sci. USA 101, 8066–8071, doi:10.1073/pnas.0308602101 (2004).
Kageyama, K. & Asano, T. Life cycle of Plasmodiophora brassicae. J. Plant Growth Regul. 28, 203–211, doi:10.1007/s00344-009-9101-z (2009).
Pageau, D., Lajeunesse, J. & Lafond, J. Impact de l’hernie des crucifères [Plasmodiophora brassicae] sur la productivité et la qualité du canola. Can. J. Plant Pathol. 28, 137–143, doi:10.1080/07060660609507280 (2006).
Voorrips, R. E. Plasmodiophora brassicae: aspects of pathogenesis and resistance in Brassica oleracea. Euphytica 83, 139–146, doi:10.1007/bf01678041 (1995).
Williams, P. H. A system for the determination of races of Plasmodiophorabrassicae that infect cabbage and rutabaga. Phytopathology 56, 624–626 (1966).
Strelkov, S. E., Tewari, J. P. & Smith-Degenhardt, E. Characterization of Plasmodiophora brassicae populations from Alberta, Canada. Can. J. Plant Pathol. 28, 467–474, doi:10.1080/07060660609507321 (2006).
Strelkov, S. E., Manolii, V. P., Cao, T., Xue, S. & Hwang, S. F. Pathotype classification of Plasmodiophora brassicae and its occurrence in Brassica napus in Alberta, Canada. J. Phytopathol. 155, 706–712, doi:10.1111/j.1439-0434.2007.01303.x (2007).
Xue, S., Cao, T., Howard, R. J., Hwang, S. F. & Strelkov, S. E. Isolation and variation in virulence of single-spore isolates of Plasmodiophora brassicae from Canada. Plant Dis. 92, 456–462, doi:10.1094/pdis-92-3-0456 (2008).
Strelkov, S. E. & Hwang, S. F. Special Issue: Clubroot in the Canadian canola crop: 10 years into the outbreak. Can. J. Plant Pathol. 36, 27–36, doi:10.1080/07060661.2013.863807 (2014).
Strelkov, S. E., Hwang, S. F., Manolii, V. P., Cao, T. & Feindel, D. Emergence of new virulence phenotypes of Plasmodiophora brassicae on canola (Brassica napus) in Alberta, Canada. Eur. J. Plant Pathol. 145, 517–529, doi:10.1007/s10658-016-0888-8 (2016).
Hasan, M. J., Strelkov, S. E., Howard, R. J. & Rahman, H. Screening of Brassica germplasm for resistance to Plasmodiophora brassicae pathotypes prevalent in Canada for broadening diversity in clubroot resistance. Can. J. Plant Sci. 92, 501–515, doi:10.4141/cjps2010-006 (2012).
Peng, G. et al. Sources of resistance to Plasmodiophora brassicae (clubroot) pathotypes virulent on canola. Can. J. Plant Pathol. 36, 89–99, doi:10.1080/07060661.2013.863805 (2014).
Johnston, T. D. Transfer of disease resistance from Brassica campestris L. to rape (B. napus L.). Euphytica 23, 681–683, doi:10.1007/bf00022490 (1974).
Gowers, S. The transfer of characters from Brassica campestris L. to Brassica napus L.: Production of clubroot-resistant oil-seed rape (B. napus ssp oleifera). Euphytica 31, 971–976, doi:10.1007/bf00039237 (1982).
Yu, F., Lydiate, D. J., Gugel, R. K., Sharpe, A. G. & Rimmer, S. R. Introgression of Brassica rapa subsp. sylvestris blackleg resistance into B. napus. Mol. Breed 30, 1495–1506, doi:10.1007/s11032-012-9735-6 (2012).
Gautam, M., Ge, X. H. & Li, Z. Y. “Brassica” In Alien Gene Transfer in Crop Plants, Volume 2: Achievements and Impacts, 207–229 (Springer New York, 2014).
Li, Q. et al. A large-scale introgression of genomic components of Brassica rapa into B. napus by the bridge of hexaploid derived from hybridization between B. napus and B. oleracea. Theor. Appl. Genet. 126, 2073–2080, doi:10.1007/s00122-013-2119-4 (2013).
Gao, F. et al. Fine mapping a clubroot resistance locus in Chinese cabbage. J. Am. Soc. Hortic. Sci. 139, 247–252 (2014).
Chen, J. et al. Identification of novel QTLs for isolate-specific partial resistance to Plasmodiophora brassicae In Brassica rapa. PLoS ONE 8, e85307, doi:10.1371/journal.pone.0085307 (2013).
Cho, K. H. et al. Mapping quantitative trait loci (QTL) for clubroot resistance in Brassica rapa L. J. Hortic. Sci. Biotechnol. 87, 325–333, doi:10.1080/14620316.2012.11512872 (2012).
Chu, M. et al. Fine mapping of Rcr1 and analyses of its effect on transcriptome patterns during infection by Plasmodiophora brassicae. BMC Genomics 15, 1166, doi:10.1186/1471-2164-15-1166 (2014).
Hayashida, N. et al. Construction of a practical SCAR marker linked to clubroot resistance in Chinese cabbage, with intensive analysis of HC352b genes. J. Jpn. Soc. Hortic. Sci. 77, 150–154, doi:10.2503/jjshs1.77.150 (2008).
Hirai, M. et al. A novel locus for clubroot resistance in Brassica rapa and its linkage markers. Theor. Appl. Genet. 108, 639–643, doi:10.1007/s00122-003-1475-x (2004).
Kato, T., Hatakeyama, K., Fukino, N. & Matsumoto, S. Fine mapping of the clubroot resistance gene CRb and development of a useful selectable marker in Brassica rapa. Breed. Sci 63, 116–124, doi:10.1270/jsbbs.63.116 (2013).
Kuginuki, Y. et al. RAPD markers linked to a clubroot-resistance locus in Brassica rapa L. Euphytica 98, 149–154, doi:10.1023/A:1003147815692 (1997).
Matsumoto, E., Yasui, C., Ohi, M. & Tsukada, M. Linkage analysis of RFLP markers for clubroot resistance and pigmentation in Chinese cabbage (Brassica rapa ssp. pekinensis). Euphytica 104, 79–86, doi:10.1023/A:1018370418201 (1998).
Pang, W. et al. Genetic detection of clubroot resistance loci in a new population of Brassica rapa. Hortic. Environ. Biotechnol 55, 540–547, doi:10.1007/s13580-014-0079-5 (2014).
Piao, Z. Y., Deng, Y. Q., Choi, S. R., Park, Y. J. & Lim, Y. P. SCAR and CAPS mapping of CRb, a gene conferring resistance to Plasmodiophora brassicae in Chinese cabbage (Brassica rapa ssp. pekinensis). Theor. Appl. Genet. 108, 1458–1465, doi:10.1007/s00122-003-1577-5 (2004).
Saito, M. et al. Fine mapping of the clubroot resistance gene, Crr3, In Brassica rapa. Theor. Appl. Genet. 114, 81–91, doi:10.1007/s00122-006-0412-1 (2006).
Sakamoto, K., Saito, A., Hayashida, N., Taguchi, G. & Matsumoto, E. Mapping of isolate-specific QTLs for clubroot resistance in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Theor. Appl. Genet. 117, 759–767, doi:10.1007/s00122-008-0817-0 (2008).
Suwabe, K. et al. Identification of two loci for resistance to clubroot (Plasmodiophora brassicae Woronin) In Brassica rapa L. Theor. Appl. Genet. 107, 997–1002, doi:10.1007/s00122-003-1309-x (2003).
Suwabe, K. et al. Simple sequence repeat-based comparative genomics between Brassica rapa and Arabidopsis thaliana: The genetic origin of clubroot resistance. Genetics 173, 309–319, doi:10.1534/genetics.104.038968 (2006).
Yu, F. et al. Identification of genome-wide variants and discovery of variants associated with Brassica rapa clubroot resistance gene Rcr1 through bulked segregant RNA sequencing. PLoS One. 11, e0153218, doi:10.1371/journal.pone.0153218 (2016).
Zhang, T. et al. Fine genetic and physical mapping of the CRb gene conferring resistance to clubroot disease in Brassica rapa. Mol. Breed 34, 1173–1183, doi:10.1007/s11032-014-0108-1 (2014).
Grandclément, C. & Thomas, G. Detection and analysis of QTLs based on RAPD markers for polygenic resistance to Plasmodiophora brassicae Woron in Brassica oleracea L. Theor. Appl. Genet. 93, 86–90, doi:10.1007/s001220050251 (1996).
Figdore, S. S., Ferreira, M. E., Slocum, M. K. & Williams, P. H. Association of RFLP markers with trait loci affecting clubroot resistance and morphological characters in Brassica oleracea L. Euphytica 69, 33–44, doi:10.1007/bf00021723 (1993).
Landry, B. S. et al. A genetic map for Brassica oleracea based on RFLP markers detected with expressed DNA sequences and mapping of resistance genes to race 2 of Plasmodiophora brassicae (Woronin). Genome 35, 409–420, doi:10.1139/g92-061 (1992).
Moriguchi, K., Kimizuka-Takagi, C., Ishii, K. & Nomura, K. A genetic map based on RAPD, RFLP, isozyme, morphological markers and QTL analysis for clubroot resistance in Brassica oleracea. Breed. Sci 49, 257–265, doi:10.1270/jsbbs.49.257 (1999).
Nagaoka, T. et al. Identification of QTLs that control clubroot resistance in Brassica oleracea and comparative analysis of clubroot resistance genes between B. rapa and B. oleracea. Theor. Appl. Genet. 120, 1335–1346, doi:10.1007/s00122-010-1259-z (2010).
Nomura, K. et al. Evaluation of F2 and F3 plants introgressed with QTLs for clubroot resistance in cabbage developed by using SCAR markers. Plant Breed. 124, 371–375 (2005).
Rocherieux, J. et al. Isolate-specific and broad-spectrum QTLs are involved in the control of clubroot in Brassica oleracea. Theor. Appl. Genet. 108, 1555–1563, doi:10.1111/j.1439-0523.2005.01105.x (2004).
Tomita, H., Shimizu, M., Asad-ud Doullah, M., Fujimoto, R. & Okazaki, K. Accumulation of quantitative trait loci conferring broad-spectrum clubroot resistance in Brassica oleracea. Mol. Breed 32, 889–900, doi:10.1007/s11032-013-9918-9 (2013).
Voorrips, R. E., Jongerius, M. C. & Kanne, H. J. Mapping of two genes for resistance to clubroot (Plasmodiophora brassicae) in a population of doubled haploid lines of Brassica oleracea by means of RFLP and AFLP markers. Theor. Appl. Genet. 94, 75–82, doi:10.1007/s001220050384 (1997).
Lee, J. et al. Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res. 23, 29–41, doi:10.1093/dnares/dsv034 (2015).
Diederichsen, E., Beckmann, J., Schondelmeier, J. & Dreyer, F. Genetics of clubroot resistance in Brassica napus ‘Mendel’. Acta Horticulturae 706, 307–312, doi:10.17660/ActaHortic.2006.706.35 (2006).
Manzanares-Dauleux, M. J., Delourme, R., Baron, F. & Thomas, G. Mapping of one major gene and of QTLs involved in resistance to clubroot in Brassica napus. Theor. Appl. Genet. 101, 885–891, doi:10.1007/s001220051557 (2000).
Werner, S., Diederichsen, E., Frauen, M., Schondelmaier, J. & Jung, C. Genetic mapping of clubroot resistance genes in oilseed rape. Theor. Appl. Genet. 116, 363–372, doi:10.1007/s00122-007-0674-2 (2008).
Zhang, H. et al. Mapping of clubroot (Plasmodiophora brassicae) resistance in canola (Brassica napus). Plant Pathol. 65, 435–440, doi:10.1111/ppa.12422 (2016).
Fredua-Agyeman, R. & Rahman, H. Mapping of the clubroot disease resistance in spring Brassica napus canola introgressed from European winter canola cv. ‘Mendel’. Euphytica 211, 201–213, doi:10.1007/s10681-016-1730-2 (2016).
Hasan, M. J. & Rahman, H. Genetics and molecular mapping of resistance to Plasmodiophora brassicae pathotypes 2, 3, 5, 6, and 8 in rutabaga (Brassica napus var. napobrassica). Genome 59, 805–815, doi:10.1139/gen-2016-0034 (2016).
Li, L. et al. A genome-wide association study reveals new loci for resistance to clubroot disease in Brassica napus. Front. Plant Sci. 7, doi:10.3389/fpls.2016.01483 (2016).
Ueno, H. et al. Molecular characterization of the CRa gene conferring clubroot resistance in Brassica rapa. Plant. Mol. Biol. 80, 621–629, doi:10.1007/s11103-012-9971-5 (2012).
Hatakeyama, K. et al. Identification and characterization of Crr1a, a gene for resistance to clubroot disease (Plasmodiophora brassicae Woronin) In Brassica rapa L. PLoS ONE 8, e54745, doi:10.1371/journal.pone.0054745 (2013).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1040, doi:10.1038/ng.919 (2011).
Drinkwater, N. R. & Gould, M. N. The long path from QTL to gene. PLoS Genet. 8, e1002975, doi:10.1371/journal.pgen.1002975 (2012).
Pfeifer, S. P. From next-generation resequencing reads to a high-quality variant data set. Heredity 118, 111–124, doi:10.1038/hdy.2016.102 (2016).
Liu, H. et al. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genomics 15, 104, doi:10.1186/1471-2164-15-104 (2014).
Li, H. et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics 16, 216, doi:10.1186/s12864-015-1424-5 (2015).
Pootakham, W. et al. Genome-wide SNP discovery and identification of QTL associated with agronomic traits in oil palm using genotyping-by-sequencing (GBS). Genomics 105, 288–295, doi:10.1016/j.ygeno.2015.02.002 (2015).
Jones, D. R., Ingram, D. S. & Dixon, G. R. Factors affecting tests for differential pathogenicity in populations of Plasmodiophora brassicae. Plant Pathol. 31, 229–238, doi:10.1111/j.1365-3059.1982.tb01273.x (1982).
Bradshaw, H. D. Jr. et al. Quantitative trait loci affecting differences in floral morphology between two species of monkeyflower (Mimulus). Genetics 149, 367–382 (1998).
Kuginuki, Y., Yoshikawa, H. & Hirai, M. Variation in virulence of Plasmodiophora brassicae in Japan tested with clubroot-resistant cultivars of Chinese cabbage (Brassica rapa L. ssp. pekinensis). Eur. J. Plant Pathol. 105, 327–332, doi:10.1023/A:1008705413127 (1999).
Horiuchi, S. & Hori, M. A simple greenhouse technique for obtaining high levels of clubroot incidence. Bulletin of the Chugoku National Agricultural Experiment Station, E, 33–55 (1980).
Iversen, G. R. & Gergen, M. Statistics: The Conceptual Approach (Springer undergraduate textbooks in Statistics) (1997).
Van ooijen, J. V. & Voorrips, R. E. Join Map® 3.0, software for the calculation of genetic linkage maps. Plant Research International, Wageningen, Netherlands (2001).
Voorrips, R. E. M. Software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78, doi:10.1093/jhered/93.1.77 (2002).
Conesa, A. et al. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676, doi:10.1093/bioinformatics/bti610 (2005).
Chalhoub, B. et al. Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345, 950–953, doi:10.1126/science.1253435 (2014).
Acknowledgements
We thank Dr. Victor Manolii (University of Alberta) and Mr. Jinghe Wang (SRDC, AAFC) for phenotyping plants for resistance to clubroot. Dr. Fuyou Fu (SRDC, AAFC) provided great help with analyzing the SNP markers with software Mapchart 2.1 and MapQTL 6. This work was funded by the Canola Agri-Science Cluster II.
Author information
Authors and Affiliations
Contributions
F.Y. and G.P. conceived this research. F.Y. designed the experiments and performed data analysis; X.Z. developed the mapping population, collected leaf samples and extracted DNA; G.P. and K.C.F. identified or developed the resistant line and provided the parental lines; S.E.S. and B.D.G. completed phenotyping; F.Y. drafted the manuscript; all authors reviewed the manuscript and approved the final draft.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, F., Zhang, X., Peng, G. et al. Genotyping-by-sequencing reveals three QTL for clubroot resistance to six pathotypes of Plasmodiophora brassicae in Brassica rapa . Sci Rep 7, 4516 (2017). https://doi.org/10.1038/s41598-017-04903-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04903-2
This article is cited by
-
Identification of QTLs for resistance to 10 pathotypes of Plasmodiophora brassicae in Brassica oleracea cultivar ECD11 through genotyping-by-sequencing
Theoretical and Applied Genetics (2023)
-
Evolutionary expansion and expression dynamics of cytokinin-catabolizing CKX gene family in the modern amphidiploid mustard (Brassica sp.)
3 Biotech (2022)
-
In silico integration of disease resistance QTL, genes and markers with the Brassica juncea physical map
Molecular Breeding (2022)
-
Development of molecular markers based on CRa gene sequencing of different clubroot disease-resistant cultivars of Chinese cabbage
Molecular Biology Reports (2022)
-
Identification of resistance loci against new pathotypes of Plasmodiophora brassicae in Brassica napus based on genome-wide association mapping
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
