
Abstract
Human parainfluenza viruses represent a leading cause of lower respiratory tract disease in children, with currently no available approved drug or vaccine. The viral surface glycoprotein haemagglutinin-neuraminidase (HN) represents an ideal antiviral target. Herein, we describe the first structure-based study on the rearrangement of key active site amino acid residues by an induced opening of the 216-loop, through the accommodation of appropriately functionalised neuraminic acid-based inhibitors. We discovered that the rearrangement is influenced by the degree of loop opening and is controlled by the neuraminic acid’s C-4 substituent’s size (large or small). In this study, we found that these rearrangements induce a butterfly effect of paramount importance in HN inhibitor design and define criteria for the ideal substituent size in two different categories of HN inhibitors and provide novel structural insight into the druggable viral HN protein.
Similar content being viewed by others


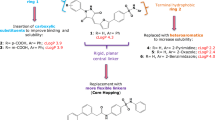
Introduction
Human parainfluenza virus (hPIV) is one of the leading causes of respiratory tract disease in infants and children1, 2 and is estimated to result in over 1.5 million cases per year in the United States alone3. Despite continuous efforts4, 5, there are neither specific antiviral therapy nor vaccines available against hPIV-3 to date. The viral surface glycoprotein haemagglutinin-neuraminidase (HN) represents an ideal target for the development of urgently needed antiviral agents. The viral HN protein encompasses three key functions in virus infection and spread. The hPIV HN recognizes and attaches to N-acetylneuraminic acid-containing glycoconjugates present on the host cell and subsequently activates the fusion machinery, facilitating infection. Upon virus replication, hPIV HN enzymatically cleaves the neuraminic acid (Neu), N-acetylneuraminic acid (Neu5Ac, 1) from host cell receptors allowing viral spread to uninfected cells3. A number of Neu2en (2)-based inhibitors (e.g. BCX 2798, 3)6, 7, as well as a novel approach that uses two well-established drugs8 in a combinatorial manner, have been used to target the hPIV-3 HN protein. However, none of these agents have progressed to the clinic.
Recently, we have reported9 the first structural investigation into the catalytic mechanism of the hPIV-3 HN protein using the 2,3-difluoro-N-acylneuraminic acid derivative, 4. This study demonstrated that the protein forms a covalent adduct with the substrate as a result of a nucleophilic attack at the Neu moiety’s anomeric carbon (C-2) by the hydroxyl group of the key catalytic amino acid, Tyr530. Hence, it was verified9 that hPIV-3 HN can be targeted by reactive substrate-like inhibitors such as 4.
We have also recently described the design and synthesis of novel potent 4-deoxy-4-triazolo-Neu2en-based inhibitors (5 and 6, Fig. 1)10. These inhibitors carry bulky C-4 substituents on the Neu2en template and target the proposed 216-cavity formed by movement of the flexible 216-loop11, a unique feature in hPIV-3 HN. Taken together these developments inspired further structural and biological investigation of Neu (1)-based inhibitors that incorporate a C-5 isobutyramido moiety, bulky C-4 triazole-substituents and C-2 and C-3 fluorides, such as 7 and 8, to evaluate functional group synergism.

Structures of reference and novel inhibitors and synthetic strategy. (a) Structures of Neu5Ac (1), reference Neu2en hPIV-3 HN inhibitors 2, 3, 5 and 6, the reference 2α,3β-difluoro-analogue of BCX 2798 (4) and the novel 2α,3β-difluoro-4-triazolo-N-acylneuraminic acid derivatives 7 and 8. (b) (i) CuSO4, sodium ascorbate, MeOH/H2O (1:1), M.W., 80 °C, 30 min (10, 81%; 11, 89%); (ii) NaOH, MeOH/H2O (1:1), rt, o/n (7, 68%; 8, 84%).
Results and Discussion
Analysis of the hPIV-3 HN structure in its open 216-loop form
To aid the development of substrate-like mechanism-based inhibitors, with further functionalization as in 5 and 6, atomic-level details of the binding mode of both 2,3-difluoro-N-acylneuraminic acid-based and Neu2en-based inhibitors were required. Therefore, both inhibitors 5 and 6 were independently co-crystallized with hPIV-3 HN protein (see Supplementary Methods).
The crystallographic structures of hPIV-3 HN in complex with inhibitors 5 (protein data bank (PDB) accession code: 5KV9) and 6 (PDB accession code: 5KV8) demonstrated that the C-4 triazole substituents occupy the 216-cavity (Fig. 2a).

Structural insights into hPIV-3 HN–5 and –6 complexes. (a) Superimposition of the hPIV-3 HN active site with bound inhibitor 5 and bound inhibitor 6 (magenta and green, respectively). A further 1.9 Å shift in 216-loop position led to the accommodation of the bulkier 4-phenyltriazole in the hPIV-3 HN–5 complex (dashed-line in black). (b) Zoom-in image displays the distances between active site residues Arg192 (side-chain) and Cys214 (backbone carbonyl oxygen) as well as the Arg192 side-chain and inhibitors 5 and 6. Dashed-lines in black or red represent favourable (H-bond donor–acceptor distance < 4.0 Å) and unfavourable distance (H-bond donor–acceptor distance > 4.0 Å) for hydrogen bonds and salt bridge formation respectively. O red, N blue.
To accommodate the C-4 substituents of each inhibitor, the respective 216-loop (amino acids 210–220)11 was forced into a more open conformation in each complex when compared with the apo structure (PDB accession code: 4XJQ). In this context, the 216-loop moved, relative to the apo structure (measured from the protein backbone), up to 3.7 Å and 1.7 Å in the hPIV-3 HN–5 and hPIV-3 HN–6 complexes, respectively. To accommodate the bulkier C-4 phenyltriazole moiety associated with inhibitor 5, the 216-loop in the hPIV-3 HN–5 complex adopted a more open conformation in comparison to the hPIV-3 HN–6 complex. The observed loop movement between these two complexes reached significant differences of up to 2.17 Å (e.g. 1.90 Å at Asp216, Fig. 2a), where the phenyltriazole moiety of 5 is directed.
Key structural differences between hPIV-3 HN–5 and HN–6 co-crystal structures
Key differences related to the degree of loop opening were observed in the position of the triarginyl cluster and the active site residues (Tyr530, Glu549, Lys254). The triarginyl cluster is known to make substantial contribution, through engagement with the carboxylate of both substrates and inhibitors, in a broad range of neuraminidases (NAs)9, 12,13,14,15 including influenza A virus (IAV) NA16,17,18,19. A similar contribution was observed in the hPIV-3 HN–6 complex, in which the hPIV-3 HN triarginyl cluster (comprised of arginine residues Arg192, Arg424 and Arg502) was found to form a salt bridge and hydrogen bonds with the carboxylic acid moiety of inhibitor 6 (Fig. 2b). Interestingly, a significant movement of the 216-loop, that led to a more open cavity in the hPIV-3 HN–5 complex, was accompanied by the reorientation of Arg192. In the hPIV-3 HN–5 complex the η2-NH2 of Arg192 side-chain, which usually participates in the coordination of the bound inhibitor, as in the hPIV-3 HN–6 complex, was oriented away from its typical location by 1.23 Å (Fig. 2b). This movement enabled the formation of a hydrogen bond between Arg192 and the Cys214 backbone carbonyl oxygen (2.59 Å). However, this shift, in the position of the Arg192 side-chain (4.22 Å, Fig. 2b) prevented a hydrogen bond formation with the carboxylic acid oxygen of inhibitor 5 and loss of a crucial ionic interaction. The two other arginine residues in the triarginyl cluster, Arg424 and Arg502, were found to maintain the same orientation in both complexes and were engaged in hydrogen bonding and ionic interactions with the carboxylate moiety of both inhibitors 5 and 6.
Further important structural differences between the two complexes were observed for the active site residues Lys254, Tyr530 and Glu549. The C-4 hydrophobic phenyltriazole moiety of inhibitor 5 is nicely accommodated in an active site groove that is lined with the side-chains of Arg192, Ile210 and Lys254 (Fig. 3a).

hPIV-3 HN–5 and –6 complexes. hPIV-3 HN active site complexed with inhibitor 5 (top) and with inhibitor 6 (bottom) displaying amino acids that are engaged in the binding event as well as three active site resident water molecules. Potential hydrogen bonds are shown as black dotted lines. O red, N blue.
Interestingly, in the hPIV-3 HN–6 complex the reorientation of the Lys254 side-chain towards the 4-methoxymethyltriazole moiety of 6 was observed. This reorientation leads to an additional hydrogen bond formation between the side-chain of Lys254 and the C-4 triazole methoxy oxygen associated with 6. In addition, the Asn208 side-chain shifts towards the side-chain amino group of Lys254, which establishes a hydrogen bond with the carbonyl oxygen of Asn208 (Fig. 3b). In stark contrast, in the hPIV-3 HN–5 complex a hydrogen bond between Asp216 and Lys254 was formed and further stabilized the active site (Fig. 3a).
The key catalytic Tyr530 residue is a highly conserved amino acid among all known wild type neuraminidases20, 21. The phenolic oxygen of Tyr530 is known to nucleophilically attack the C-2 anomeric carbon of a bound neuraminic acid derivative, as previously described9 for the 2,3-difluoroneuraminic acid-based derivative 4. Accordingly, in the hPIV-3 HN–5 and hPIV-3 HN–6 complexes the Tyr530 phenolic oxygen was found at a distance of 2.9 Å and 2.7 Å from the C-2 of inhibitors 5 and 6, respectively. The structure of the co-crystallized complex with inhibitor 6 had two protein molecules per asymmetric unit (binding site A and B), whereas the structure of the co-crystallized complex with compound 5 had only one protein molecule per asymmetric unit (see Supplementary Methods). Two orientations of Tyr530 were found in binding site A of the hPIV-3 HN–6 complex: One orientation of the residue Tyr530 was directed towards the binding site (more populated, 52%) and in the second, the residue was oriented away from the binding site. In contrast, in binding site B, Tyr530 was only directed towards the C-2 of inhibitor 6. No further differences were observed between binding site A and B of the hPIV-3 HN–6 complex.
Glu549 is one of the seven highly-conserved amino acids in the HN’s active site21, and was found oriented away from the binding site in the hPIV-3 HN–5 complex. In contrast, the Glu549 residue in the hPIV-3 HN–6 complex was always directed towards the binding site to form a hydrogen bond with an active site-bound water. Interestingly, in the recently described hPIV-3 HN–4 complex9, Glu549 is also found to orient only towards the binding site.
Effect of the triarginyl cluster shift on inhibitor complexation to the HN protein
The reaction mechanism of the broad NA family has been the subject of intense investigation over the last twenty years13,14,15, especially the IAV NA mechanism16, 18, 19, 22, 23. The triarginyl cluster–Neu5Ac carboxylate interaction is thought to have a major influence on the geometry of 1 when bound within the active site23. While the Neu2en inhibitors 5 and 6 have planar geometry around C-2 and C-3, as a consequence of their sp2-hybridization, 2,3-difluorinated Neu derivatives such as 7 and 8 need to planarise around these carbon atoms upon binding to the enzyme for the catalytic reaction to proceed19. As a consequence of the 216-loop movement due to the presence of a C-4 bulky substituent, as seen in the hPIV-3 HN–5 complex, the induced side-chain rearrangement of Arg 192 results in the loss of a crucial interaction with the carboxyl group of inhibitor 5. Hence, the introduction of bulky C-4 substituents, such as a phenyltriazole moiety, in 2,3-difluoro Neu substrate-based derivatives (e.g. 7), may in fact be counterproductive and result in poor inhibition of hPIV-3 HN. Conversely, the structure of hPIV-3 HN in complex with inhibitor 6 revealed that the Arg192 side chain remains in close proximity to the inhibitor’s carboxyl group, leading to the expected interactions (Fig. 2b). This structural observation suggests that a 2,3-difluoro derivative of 6 that incorporates a smaller C-4 functionality (e.g. 8), may undergo efficient planarization upon complexation with HN. This planarization should readily promote the expected nucleophilic attack at C-2 of 8 by the phenolic oxygen of Tyr530 to provide the commensurate β-tyrosinyl neuraminide (see Supplementary Figure 2). Finally, this should result in a complex that is similar to what has been previously observed in the recent X-ray crystal structure of the hPIV-3 HN–4 complex (PDB accession code: 4XJR)9 and consequently in a superior antiviral potency, as seen for 4.
Synthesis and HN enzyme activity evaluation of the 2,3-difluoro analogues of inhibitors 5 and 6
To investigate these hypotheses, the 2,3-difluoro analogues 7 and 8 of the potent hPIV-3 inhibitors 5 and 6 were synthesized as shown in Fig. 1b (see Supplementary Methods). Subsequently, neuraminidase inhibition (NI) IC50 values were measured by the standard NI fluorescence assay based on the method of Potier et al.24 using 4-methylumbelliferyl N-acetylneuraminide (MUN) as substrate (Fig. 4a)10. A 2-fold increase in potency was observed for the novel 2,3-difluoro inhibitor 8 that incorporates the smaller C-4 methoxymethyl triazole substituent (NI IC50 = 6.7 µM) compared to its Neu2en analogue (6, NI IC50 = 14.5 µM). In complete contrast, and as predicted from the analysis of the hPIV-3 HN–5 structure, a significant loss in potency against hPIV-3 HN neuraminidase function was observed for inhibitor 7 (NI IC50 = 55.3 µM compared with an IC50 = 1.4 µM for the Neu2en-based inhibitor 5).

Biological evaluation of the 2,3-difluoro inhibitors in relation to their Neu2en analogues. (a) Inhibitors 5, 6, 7, and 8 were evaluated in a neuraminidase assay. These values are the means of determinations from 3 independent experiments performed in duplicates and error bars correspond to calculated standard deviations. (b) Virus growth inhibition was determined by an in situ ELISA technique using the human cell line A549. IC50 values of 11.2 ± 0.9 µM, 40.1 ± 7.0 µM, 82.4 ± 7.7 µM and 14.3 ± 0.3 µM were determined for 5, 7, 6 and 8, respectively. These values were evaluated from at least 2 independent experiments performed in triplicate and error bars correspond to the calculated standard deviation.
Inhibition of haemagglutinin function of hPIV-3 HN by 5 and 6 and their 2,3-difluoro derivatives (7 and 8)
Since hPIV-3 HN is a unique multifunctional protein, that possesses both carbohydrate-binding and carbohydrate-processing functionalities in one binding site25, we were also interested to explore how difluorination of the inhibitors can affect the hPIV-3 HN haemagglutinin function. For this purpose, virus-mediated agglutination of human red blood cells was evaluated in the presence of decreasing inhibitor concentrations (see Supplementary Methods) in a similar procedure to that previously reported10. Results from a haemagglutinin inhibition (HI) assay demonstrated a greatly enhanced potency for the non-fluorinated inhibitors 5 and 6 when compared to their fluorinated analogues 7 and 8 (see Supplementary Figure 3). These significant weaker HI IC50 values of the 2,3-difluoro derivatives are in agreement with a recently reported 2,3-difluoro-N-acylneuraminic acid26 that has a HI IC50 value of 1 mM and the biotinylated oligosaccharide, 3′SiaDiLN (Neu5Acα2–3 Galβ1–4GlcNAcβ1–3 Galβ1–4GlcNAc)27 that has a K d value of 4.3 mM when in complex with soluble hPIV-3 HN. The Neu2en-based inhibitors 5 and 6 are significantly more potent in HI with IC50 values of 0.6 µM and 3.9 µM, respectively. Therefore, it seems that planar geometry around C-2 and C-3 of the Neu2en template has an influence on the extent of hPIV-3 HN haemagglutinin function inhibition. Interestingly, the stronger HI for 8 (IC50 = 57 µM) when compared to 7 (IC50 = 775 µM) at 4 °C may be primarily attributed to the ability of inhibitor 8 to engage the triarginyl cluster and the failure of inhibitor 7 to bind to Arg192, and secondly to the formation of an additional hydrogen bond interaction between the Lys254 side-chain and the C-4 methoxymethyl oxygen of 8.
Blockade of hPIV-3 virus propagation in cells by inhibitors 5–8
To further investigate the impact of the C-4 substituent size of the inhibitors on hPIV-3 growth, probes 5–8 were evaluated at a cellular level for their capacity to block virus infection and propagation in the human respiratory cell line A549 (lung adenocarcinoma epithelial cells) using our well-established in situ ELISA technique10. The 2,3-difluoro derivative 8 with the relatively smaller C-4 substituent showed an enhanced capacity to inhibit virus growth, with an experimentally–determined IC50 value of 14.3 µM compared to 82.4 µM for the comparable Neu2en inhibitor 6. This is consistent with the IC50 value determinations from NI assay data and is similar to what is observed9 for inhibitor 4 with a smaller C-4 azido functionality. Since the C-4 substituents of 4 and 8 are not bulky enough to induce significant movement of the 216-loop, Arg192 remains in its original orientation that maintains the integrity of the triarginyl cluster–Neu carboxylate interactions while maintaining its hydrogen bond to the Cys214 residue (Fig. 2b). In complete agreement with NI enzyme assay data, inferior potency in the virus growth inhibition assay was observed for inhibitor 7 that has the bulkier C-4 phenyltriazole substituent. IC50 values of 40.1 µM and 11.2 µM were determined for 7 and its Neu2en analogue 5, respectively (Fig. 4a).
Finally, all inhibitors were assessed for cytotoxicity to human A549 cells with no toxicity observed at a concentration of 300 µM (data not shown). In addition, we investigated9 the capacity of the synthesized inhibitors 5–8 to inhibit human neuraminidase 2 (hNeu2) compared with the well-described neuraminidase inhibitor Neu5Ac2en (2)28. No inhibition of hNeu2 was observed for either the 2,3-difluoro-N-acylneuraminic acid-based inhibitors (7 & 8) or their corresponding Neu2en analogues (5 & 6) (see Supplementary Figure 4).
Conclusion
In conclusion, we provide the first structural insights of the hPIV-3 HN protein in a 216-loop open form and describe an in-depth analysis of the rearrangement of binding pocket-resident amino acids as a result of this movement. We have also demonstrated that there is a direct relationship between inhibitor size and the inhibitor’s capacity to open the loop without losing the key interactions between the important active site-based triarginyl cluster and inhibitor. The additional complexity of loop flexibility can directly impact the extent of inhibitor functionalization. In the case of hPIV-3 HN whether large or small C-4 substituents are optimal on the described inhibitors, will depend on the inhibitor framework. From our study “bigger is better” when using the Neu2en framework as the loss of the ionic interaction with Arg192 is compensated by the affinity gain from the hydrophobic C-4 moiety that is oriented in the 216-cavity. In contrast, “smaller is better” when using the 2,3-difluoro-Neu5Ac framework, as these inhibitors are designed to react with the active site Tyr530, thus targeting the catalytic machinery of HN. Smaller substituents at C-4 do not have significant impact on the 216-loop or induce amino acid rearrangement. Consequently, the triarginyl cluster residue Arg192 maintains the capacity to interact with the inhibitor’s carboxylate moiety and facilitates planarization of the substrate-like inhibitor to optimally position C-2 of the difluoro inhibitors for nucleophilic attack by Tyr530.
This study has shown that hPIV-3 HN is a complex flexible protein. The protein’s flexibility, in the context of inhibitor development can be considered a butterfly effect, since a small structural change in one part of the protein has a significant influence on another HN region. This challenge has been recently recognized by the computational biology community29. As a consequence, optimal anti-parainfluenza virus drug design that accounts for the hPIV-3 HN protein flexibility and inhibitor-associated structural rearrangements is required. Targeting multiple individual structural features may not lead to the expected synergistic effect, if done without consideration of the impact of structural changes occurring at the whole protein level, and consequently on bound inhibitors. Finally, our findings define an important set of criteria that reveal an critical balancing act with new essential parameters that will assist in the design of novel hPIV-3 HN inhibitors.
Material and Methods
General Methods Chemistry
Commercially available reagents and dry solvents were purchased from commercial sources and used without further purification. Anhydrous reactions were carried out using oven-dried glassware and under an argon atmosphere. Reaction progress was monitored through thin layer chromatography (TLC) using aluminium plates pre-coated with Silica Gel 60 F254 (E. Merck). TLC plates were observed under UV light at 254 nm and were then visualized by application of a solution of H2SO4 in EtOH (5% v/v) followed by heating. Flash chromatography was carried out using Silica Gel 60 (0.040–0.063 mm) and distilled solvents. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively on a BrukerAvance 400 MHz spectrometer, while 19F NMR spectra were recorded at 376 MHz. Chemical shifts (δ) are reported in parts per million, relative to the internal reference residual solvent peak [CDCl3: 7.26 (s) for 1H, 77.0 (t) for 13C; D2O: 4.79 (s) for 1H]. 2D COSY and HSQC experiments were run when necessary to support assignments. Low-resolution mass spectra (LRMS) were recorded using positive mode, in electrospray ionization mode, on a BrukerDaltonics Esquire 3000 ESI spectrometer. High-resolution mass spectra (HRMS) were recorded for the final derivatives using Griffith University SmartWater Research Centre facility on Agilent 6530 QTOF, with an Agilent 1290 HPLC. Final deprotected derivatives were purified by reversed phase chromatography, using GracePureTM SPE C18-Aq 5000 mg/20 mL and 2% acetonitrile/H2O. The purities of all synthetic intermediates after chromatographic purification were > 90% as determined by 1H and 13C NMR, while the purity of reference compounds synthesized for screening purposes 5 and 6, and of the new final products 7 and 8 was judged to be ≥ 95%. The synthesis of intermediate 9 (ref. 9) and reference inhibitors 5 and 6 (ref. 10) followed typical literature procedures. The details of synthetic methods used and full characterization data of key intermediates and novel final products are reported here in the supplementary material.
General procedure for the synthesis of compounds 10 and 11
The 4-azido-4-deoxy-2α,3β-difluoro-5-isobutyramido neuraminic acid intermediate 9 (60 mg, 0.12 mmol) and the corresponding ethynyl derivative (0.24 mmol) were dissolved in a (1:1) mixture of methanol and H2O (2 mL). Copper(II)sulfate pentahydrate (6.5 mg, 0.024 mmol) was added to the mixture followed by sodium ascorbate (0.1 mL of freshly prepared 1 M solution in H2O). The mixture was heated in a microwave reactor at 80 °C for 30 min. The reaction solvent was then evaporated under vacuum, and the residue was taken up in dichloromethane (50 mL), washed with 10% NH4OH (10 mL), followed by brine (10 mL). The organic layer was dried over anhydrous Na2SO4, concentrated in vacuo to give the crude product 10 or 11 and finally purified by silica gel chromatography using a suitable solvent system.
General procedure for the synthesis of compounds 7 and 8
To compound 10 or 11 (0.068 mmol) in a (1:1) mixture of water and MeOH (2 mL) at 0 °C was added an aq. solution of NaOH (1.0 M) dropwise until a pH of 13–14 was reached. The reaction mixture was allowed to warm up gradually to rt and was stirred at rt overnight. The solution was then rendered acidic with Amberlite® IR-120 (H+) resin (to pH = 5), filtered through a cotton plug and washed with MeOH (10 mL) and H2O (10 mL). The compound was then purified by reversed phase chromatography, using C18-GracePureTM cartridge and 2% acetonitrile/water, to yield pure 7 (28 mg, 84%) or 8 (21 mg, 68%), as a fluffy white powder after freeze drying.
Cells and viruses
LLC-MK2 (monkey kidney epithelial cells) cells were maintained at 37 °C, 5% CO2 in EMEM (Lonza, Basel, Switzerland) supplemented with 1% Glutamine (1% final concentration) and 2% heat-inactivated fetal bovine serum (FBS). A549 cells (human lung adenocarcinoma epithelial cells) were provided by the European Collection of Cell Cultures (86012804–1VL, Sigma-Aldrich). Cells were propagated in Dulbecco’s Modified Eagle Medium (DMEM) (Lonza, Basel, Switzerland) supplemented with of 1% Glutamine and 5% FBS. For infection procedures, A549 cells were maintained in DMEM supplemented with 1% Glutamine only.
hPIV-3 (strain C-243) was purchased from the American Type Culture Collection (Manassas, VA) and virus stock was amplified in LLC-MK2 cells at 35 °C, 5% CO2 in EMEM (only supplemented 1% Glutamine). After 96 hours of incubation, the virus-containing supernatant was harvested, clarified and purified by PEG precipitation followed by sucrose gradient fractionation as previously published10.
hPIV-3 HN inhibitors
Compounds 2–8 were each provided as a lyophilized powder and then reconstituted in sterile water to generate a 10 mM stock solution. Solutions were sonicated for 15 min to allow complete dissolution and then filter-sterilized. The stock solution was stored in a glass vial at −20 °C and freshly diluted in appropriate buffer before usage.
Recombinant human Neuraminidase 2 (hNeu2) expression and purification
The entire coding sequence of hNeu2 was cloned into pGEX-2T expression vector (Amersham Biosciences). Recombinant hNeu2 expression was performed in E. Coli cells. Purification of recombinant hNeu2 was accomplished in three steps using affinity chromatography, followed by two successive steps of high resolution anion and cation exchange chromatography as previously described12, 30.
Neuraminidase inhibition assay
Viral neuraminidase inhibition (NI) assays were accomplished with purified hPIV-3 in reaction buffer/hPIV3 (NaOAc 50 mM, CaCl2 5 mM, pH 4.6). The maximal fluorescence signal of purified virus (positive control) was required to be at least 5 times higher than the fluorescence background (negative control) in order to be considered as statistically relevant. NI assays were performed in triplicate following the protocol as described earlier9, 10.
The NI assay with human Neuraminidase 2 (hNeu2) was performed as previously described9, using 60 ng of purified hNeu2 enzyme in reaction buffer/hNeu2 (NaOAc 50 mM, pH 5.5), supplemented with 10 μg BSA and 0.5 mM MUN per reaction. All inhibitors were tested in duplicate in the concentration range from 0.001 μM to 1000 μM. After 10 min of incubation with agitation (1000 rpm) at 37 °C, the enzymatic reaction was stopped with glycine buffer (glycine 0.25 M, pH 10.4) and the product fluorescence was measured in relative fluorescence units (RFU) using a Victor 3 multilabel reader (PerkinElmer, Waltham, MA). Controls were conducted with MUN and purified hNeu2, the enzymatic reaction was stopped at t = 0 for the negative control and at t = 10 min for the positive control.
The obtained data were normalised by background (negative control RFU) subtraction and processed data were analyzed with GraphPadPrism 4 (GraphPad Software Inc., La Jolla, CA) to calculate IC50 values. The concentration of inhibitor that reduced the neuraminidase activity by 50% compared to those of a non-treated virus/protein suspension was considered as the NI IC50 value.
Haemagglutination inhibition assay
The HN inhibitors were assessed in haemagglutination inhibition assays using human red blood cells (RBC) as previously described10 with minor modifications. The lowest final concentration of hPIV-3 that leads to a complete agglutination of human-RBC at 4 °C is considered as 1 HAU. 4 haemagglutination units (HAU) of hPIV-3 (final 1 HAU per well) were pre-mixed with inhibitors of each tested concentration (4X dilution in PBS) on ice. Next, an equivalent volume of ice-cold 1% human-RBC in PBS was added to each well of the inhibitor/virus pre-mixture and the plate was then incubated for 90 min at 4 °C before determining the extent of haemagglutination inhibition. The observed agglutination in a control well containing only 0.5 HAU of hPIV-3 and human-RBC was used to determine the concentration of the inhibitor that reduced the haemagglutination activity (agglutination) by 50% compared to those of a non-treated virus suspension. A similar observed agglutination as in the control well (0.5 HAU of hPIV-3) was considered as the HI IC50 value for each respective inhibitor.
In situ ELISA
This technique was extensively used in previous studies9, 10, to assess dose-dependent potency of compounds to inhibit virus growth by measuring expression levels of hPIV-3 HN at the cell surface of an infected cell monolayer. This method is shortly summarized here; infection was performed with 100 FFU/well of hPIV-3 for 1 h at 37 °C with gentle agitation every 15 min on a confluent A549 monolayer seeded in a 96 well-plate. Assays were performed in triplicate; inocula were removed and replaced with respective compound dilutions (final concentration of 0.001 μM to 1000 μM). Virus proliferation was allowed at 37 °C, 5% CO2 for 36–40 h. Virus was inactivated and cells were fixed by treatment with 4% paraformaldehyde in PBS for 20 min. Next, 0.3% H2O2/PBS (final concentration) was added to inactivate endogenous peroxidases at 37 °C for 30 min. 1:2000 dilution of mouse monoclonal IgG anti-hPIV-3HN (Fitzgerald, clone# M02122321, 2.0 mg/mL) in 5% milk/PBS was incubated with the cells for 1 h at 37 °C, followed by an incubation of 1:2000 dilution of goat anti- Mouse-IgG(H + L)-HRP conjugate (BioRad, ref# 170–6516) in 5% milk/PBS for 1 h at 37 °C. BD OptEIATMB substrate (BD Biosciences, San Jose, CA, 100 μL) was added to virus-infected cells and the reaction was stopped after 3–5 min by addition of 1 M H2SO4 (50 μL). The initial absorbance (OD) of each well was read at 450 nm for 0.1 s using a Victor 3 multilabel reader. The negative control (non-infected cells) OD was subtracted from the initial ODs and final OD values were analyzed with GraphPadPrism4 to calculate IC50 values. The IC50 value was considered as the concentration of inhibitor that reduced the absorbance at 450 nm by 50% compared to a non-treated infected cell monolayer.
Cytotoxicity assay
The cytotoxcity of all compounds, described in cell-based infection assays, was assessed by incubating the inhibitors at two different concentrations (30 µM and 300 µM) with the human cell line A549 for 48 h. The cell viability was measured by adding AlamarBlue® (life technologies) reagent as 10% of the sample volume to the cells and incubate them for 4 h at 37 °C, following fluorescence was read at 570 nm with a BioRad plate reader.
Recombinant HN expression and purification
The ectodomain of the haemagglutinin-neuraminidase protein from human parainfluenza virus type 3 Washington/1957 C243 was expressed using the Bac-to-Bac® baculovirus expression system (Invitrogen, Carlsbad, CA). The nucleotide sequence for a honeybee melittin signal peptide (HBM) was added downstream to the sequence encoding for the HN ectodomain (amino acids 125 to 572). This sequence (HBM + HN) was codon optimised for expression in Spodoptera frugiperda cells (Sf9) and ordered directly through the DNA2.0 gene synthesis service (DNA2.0, Menlo Park, CA) as a gene named HBM-HNhPIV-3opt. HBM-HNhPIV-3opt was amplified by PCR and ligated into a pFastBac⁄CT-TOPO® vector that provides an additional C-terminal 6-histidine tag (His-Tag) for purification and detection purposes. The generation and amplification of recombinant baculovirus containing HBM-HNhPIV-3opt were performed according to the manufacturer’s instructions. Sf9 cells (Invitrogen), cultured in Insect-XPRESS protein free insect cell medium (Lonza), were infected with high MOI of HBM-HNhPIV-3opt baculovirus. Four days post-infection the supernatant, containing recombinant HN, was collected to yield the highest protein expression. The supernatant was clarified by centrifugation (3000 RCF for 15 min) to remove cell debris and then purified on a HisTrap excel 5 mL column (GE Healthcare life sciences, Buckinghamshire, England) following the manufacturer’s protocol. Recombinant HN was eluted with 500 mM imidazole solution and collected fractions were assessed by a neuraminidase activity (NA) assay (see below). The most active fractions were pooled and concentrated with a 10 kDa Amicon Ultra filter unit (Millipore) to a final volume of 800 μL. An additional purification step was performed that employed fast protein liquid chromatography (Amersham Biosciences) over a Superdex 75 gel filtration column (GE Healthcare) at 4 °C and 1 mL fractions were collected with a Frac-920. Protein-containing fractions, as determined by monitoring fraction collection at 280 nm, were assessed in a NA assay as well as subjected to SDS-PAGE. Purified and concentrated recombinant HN protein was stored at 4 °C.
Crystallization of hPIV-3 HN co-complexes
The 4 mg/mL hPIV-3 HN protein stock solution was pre-incubated with a final concentration of 1 mM inhibitor 5 or 1.5 mM inhibitor 6 in 0.1 M citrate buffer pH 4.6, 0.2 M ammonium sulfate and 15% PEG 3000. Crystallization trials were set up as 2 µL pre-incubated stock solution using the hanging drop vapour diffusion method. The drop was equilibrated against a 500 µL reservoir (0.1 M citrate buffer pH 4.6, 0.2 M ammonium sulfate and 15% PEG 3000). The crystals were mounted in nylon loops (Hampton Research) and flash frozen at 100 K in a cryoprotectant solution containing 20% glycerol in addition to the precipitant solution.
Data collection and structure determination
X-ray diffraction data were collected at the Photon Factory AR-NW12A31 and BL5A beam lines (Japan) at a wavelength of 1.000 Å. Images for the complex hPIV-3 HN-5 were recorded on an ADSC Q315r CCD detector using 5 s exposure per 0.5° oscillation angles. In case of hPIV-3 HN-6, images were recorded on an ADSC Q210r CCD detector using a 5 s exposure per 1° oscillation angles. The data were reduced and scaled using XDS32 and CCP4 (ref. 33) program suites. The hPIV-3 HN co-complexes were determined by molecular replacement using Phaser34 from the Phenix suite35 employing the atomic coordinates of hPIV-3 HN (PDB ID: 4XJQ) as the search model. Coot36, Refmac537 and Phenix.refine35 were employed for initial restrained, model-building and further cycles of refinement. The final models were confirmed and validated through the Molprobity38 server. X-ray data collection statistics are summarized in Supplementary Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the accession codes 5KV8 and 5KV9 for the hPIV-3 HN–5 and hPIV-3 HN–6 complex structures, respectively.
Structures of co-crystallized complexes with inhibitor 6 belonged to the P212121 space-group with unit cell dimensions of 81.57 Å × 98.61 Å × 103.39 Å and had two protein molecules per asymmetric unit as previously described for the apo-hPIV-3 HN structure (PDB accession code: 4XJQ)9 and for all reported hPIV-3 HN complexes to date9, 21, 26, 39. In contrast, for the first time, structures of co-crystallized complexes with compound 5 were found to belong to the C2221 space-group with unit cell dimensions of 80.10 Å × 107.37 Å × 94.49 Å and one protein molecule per asymmetric unit was observed. The structure of the C2221 crystal form was determined by the molecular replacement method using a monomer of apo-hPIV3-HN (PDB accession code: 4XJQ) as a search model. Analysis of the P212121 and C2221 crystal forms revealed that the overall packing of molecules are the same, but ligand induced conformational differences in the active site region of hPIV3-HN lead to a change in the symmetry axis between the molecules in the protein biological dimer from non-crystallographic in the apo form to crystallographic in the ligand-bound form resulting in a C2221 space-group.
References
Steffens, A., Finelli, L., Whitaker, B. & Fowlkes, A. Population-based Surveillance for Medically Attended Human Parainfluenza Viruses From the Influenza Incidence Surveillance Project, 2010–2014. Pediatr. Infect. Dis. J. 35, 717–722 (2016).
Abedi, G. R. et al. Estimates of Parainfluenza Virus-Associated Hospitalizations and Cost Among Children Aged Less Than 5 Years in the United States, 1998–2010. J. Pediatr. Infect. Dis. Soc 5, 7–13 (2016).
Henrickson, K. J. Parainfluenza Viruses. Clin. Microbiol. Rev. 16, 242–264 (2003).
Englund, J. A. et al. Safety and infectivity of two doses of live-attenuated recombinant cold-passaged human parainfluenza type 3 virus vaccine rHPIV3cp45 in HPIV3-seronegative young children. Vaccine 31, 5706–5712 (2013).
Moscona, A. et al. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J. Infect. Dis. 202, 234–241 (2010).
Alymova, I. V. et al. Efficacy of novel hemagglutinin-neuraminidase inhibitors BCX 2798 and BCX 2855 against human parainfluenza viruses in vitro and in vivo. Antimicrob. Agents Chemother. 48, 1495–1502 (2004).
Alymova, I. V. et al. Efficacy of the novel parainfluenza virus hemagglutinin-neuraminidase inhibitor BCX 2798 in mice—Further evaluation. Antivir. Ther. 14, 891–898 (2009).
Bailly, B. et al. A dual drug regimen synergistically blocks human parainfluenza virus infection. Sci. Rep 6, 24138 (2016).
Dirr, L. et al. The catalytic mechanism of human parainfluenza virus type 3 haemagglutinin-neuraminidase revealed. Angew. Chem. Int. Ed. 54, 2936–2940 (2015).
Guillon, P. et al. Structure-guided discovery of potent and dual-acting human parainfluenza virus haemagglutinin–neuraminidase inhibitors. Nat. Commun. 5, (2014).
Winger, M. & von Itzstein, M. Exposing the flexibility of human parainfluenza virus hemagglutinin-neuraminidase. J. Am. Chem. Soc. 134, 18447–18452 (2012).
Chavas, L. M. G. et al. Crystal structure of the human cytosolic sialidase Neu2 evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 280, 469–475 (2005).
Watts, A. G., Oppezzo, P., Withers, S. G., Alzari, P. M. & Buschiazzo, A. Structural and kinetic analysis of two covalent sialosyl-enzyme intermediates on trypanosoma rangeli sialidase. J. Biol. Chem. 281, 4149–4155 (2006).
Newstead, S. L. et al. The structure of clostridium perfringens NanI sialidase and its catalytic intermediates. J. Biol. Chem. 283, 9080–9088 (2008).
Buchini, S. et al. Tuning Mechanism-Based Inactivators of Neuraminidases: Mechanistic and Structural Insights. Angew. Chem. Int. Ed. 53, 3382–3386 (2014).
Taylor, N. R. & von Itzstein, M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J. Med. Chem. 37, 616–624 (1994).
von Itzstein, M. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 6, 967–974 (2007).
Kim, J.-H. et al. Mechanism-based covalent neuraminidase inhibitors with broad-spectrum influenza antiviral activity. Science 340, 71–75 (2013).
Vavricka, C. J. et al. Influenza neuraminidase operates via a nucleophilic mechanism and can be targeted by covalent inhibitors. Nat. Commun. 4, 1491 (2013).
Taylor, G. Sialidases: structures, biological significance and therapeutic potential. Curr. Opin. Struct. Biol. 6, 830–837 (1996).
Lawrence, M. C. et al. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335, 1343–1357 (2004).
Pascolutti, M., Thomson, R. J. & von Itzstein, M. in Methods and Principles in Medicinal Chemistry: Lead Structure Discovery (ed. Holenz, J.) (Wiley VCH, 2015).
Varghese, J. N., McKimm-Breschkin, J. L., Caldwell, J. B., Kortt, A. A. & Colman, P. M. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins. 14, 327–332 (1992).
Potier, M., Mameli, L., Bélisle, M., Dallaire, L. & Melançon, S. B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-D-N-acetylneuraminate) substrate. Anal. Biochem. 94, 287–296 (1979).
Moscona, A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 115, 1688–1698 (2005).
Streltsov, V. A., Pilling, P., Barrett, S. & McKimm-Breschkin, J. L. Catalytic mechanism and novel receptor binding sites of human parainfluenza virus type 3 hemagglutinin-neuraminidase (hPIV3 HN). Antiviral Res. 123, 216–223 (2015).
Tappert, M. M., Porterfield, J. Z., Mehta-D’Souza, P., Gulati, S. & Air, G. M. Quantitative comparison of human parainfluenza virus hemagglutinin-neuraminidase receptor binding and receptor cleavage. J. Virol. 87, 8962–8970 (2013).
Chavas, L. M. G. et al. Complexity in influenza virus targeted drug design: Interaction with human sialidases. J. Med. Chem. 53, 2998–3002 (2010).
Antunes, D. A., Devaurs, D. & Kavraki, L. E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov 10, 1301–1313 (2015).
Tringali, C. et al. Properties of recombinant human cytosolic sialidase hsNeu2 the enzyme hydrolyzes monomerically dispersed GM1 ganglioside molecules. J. Biol. Chem. 279, 3169–3179 (2004).
Chavas, L. M. G. et al. Beamline AR-NW12A: high-throughput beamline for macromolecular crystallography at the Photon Factory. J. Synchrotron Radiat. 19, 450–454 (2012).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. A graphical user interface to the CCP 4 program suite. Acta Crystallogr. D Biol. Crystallogr 59, 1131–1137 (2003).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004).
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21 (2010).
Xu, R. et al. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. mBio 4, e00803–13 (2013).
Acknowledgements
The Australian Research Council (DP1094549) and the National Health and Medical Research Council (1047824 and 1071659) are gratefully acknowledged for its financial support. Griffith University is thanked for the award of a Postdoctoral Award (IMED) and Postgraduate Scholarships (L.D.). The X-ray data was collected at the Photon Factory, Japan (Proposal No. 2012G545) and we thank T. Senda and F.X. Gallat for hosting L.D.
Author information
Authors and Affiliations
Contributions
M.v.I., P.G. and I.E-D. supervised the project. L.D. performed biological assays and crystallographic studies. I.E-D. synthesised compounds. L.D. and L.M.G.C. collected the X-ray data sets. M.v.I. and L.D. prepared the manuscript, which was reviewed by all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dirr, L., El-Deeb, I.M., Chavas, L.M.G. et al. The impact of the butterfly effect on human parainfluenza virus haemagglutinin-neuraminidase inhibitor design. Sci Rep 7, 4507 (2017). https://doi.org/10.1038/s41598-017-04656-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04656-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
