
Abstract
Deciphering the animal mitochondrial genome (mitogenome) is very important to understand their molecular evolution and phylogenetic relationships. In this study, the complete mitogenome of Clistocoeloma sinensis was determined. The mitogenome of C. sinensis was 15,706 bp long, and its A+T content was 75.7%. The A+T skew of the mitogenome of C. sinensis was slightly negative (−0.020). All the transfer RNA genes had the typical cloverleaf structure, except for the trnS1 gene, which lacked a dihydroxyuridine arm. The two ribosomal RNA genes had 80.2% A+T content. The A+T-rich region spanned 684 bp. The gene order within the complete mitogenome of C. sinensis was identical to the pancrustacean ground pattern except for the translocation of trnH. Additionally, the gene order of trnI-trnQ-trnM in the pancrustacean ground pattern becomes trnQ-trnI-trnM in C. sinensis. Our phylogenetic analysis showed that C. sinensis and Sesarmops sinensis cluster together with high nodal support values, indicating that C. sinensis and S. sinensis have a sister group relationship. The results support that C. sinensis belongs to Grapsoidea, Sesarmidae. Our findings also indicate that Varunidae and Sesarmidae species share close relationships. Thus, mitogenomes are likely to be valuable tools for systematics in other groups of Crustacea.
Similar content being viewed by others


Introduction
Mitochondrial DNA (mtDNA) is a typically closed circular molecule approximately ranging in size from 14 to 18 kb. It contains 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and control region (CR)1, 2. mtDNA is characterized by maternal inheritance, simple structure, a small genome size, conserved gene content and organization, high mutation rate, and accelerated rate of nucleotide substitution3,4,5,6,7. The mitogenomes of animal mtDNA can provide important information on rearrangement laws and phylogenetic analysis because of their rapid evolutionary rate and lack of genetic recombination1. It is becoming increasingly common to use complete animal mitogenomes for phylogenetic reconstruction8,9,10. Partial DNA sequences are often too short to contain sufficient phylogenetic information11, and combination of mitochondrial and nuclear genomes makes model selection difficult12. Further, the addition of rRNA makes alignment ambiguous13.
The infraorder Brachyura contains about 7000 described species in 98 families14. C. sinensis is one of the most important Brachyura species, and is used as a good indicators of environmental changes and water pollutions in China15. Although C. sinensis was described over 80 years ago16, it is still very poorly understood. Earlier studies classified C. sinensis into Grapsidae, Sesarminae17. In recent years, some researchers have classified C. sinensis into Grapsoidea, Sesarmidae18. Gene rearrangements in mitogenomes are useful in reconstruction of Brachyuran phylogeny19. In the present study, we sequenced the complete mitogenome of C. sinensis with the aim of elucidating its evolutionary status and rearrangement information by comparing it with complete Brachyuran mitogenomes available to date20, 21. This information may provide insights into phylogenetic rearrangement and enable phylogenetic analysis.
Methods
Sample and DNA Extraction
Adult specimens of C. sinensis were captured from Yancheng, Jiangsu province, China. Total genomic DNA was isolated from individual specimens using the Aidlab Genomic DNA Extraction Kit (Beijing, China). All procedures were completed following the manufacturer’s instructions. The complete mitogenome was amplified from the DNA from one C. sinensis crab.
PCR Amplification and Sequencing
The complete mitogenome was obtained using a combination of conventional PCR and long PCR to amplify overlapping fragments spanning the whole mitogenome. Universal and specific primers were designed based on the conserved nucleotide sequences of known mitochondrial sequences in Brachyura (Table 1) and synthesized by Beijing Sunbiotech22,23,24,25,26. The fragments were amplified using Aidlab Red Taq (Beijing, China) according to the manufacturer’s instructions. All amplifications were performed on an Eppendorf Mastercycler and Mastercycler gradient in 50 µl reaction volumes with 5 µl 10 × Taq Buffer (Mg2+) (Aidlab), 4 µl of dNTPs (2.5 mM, Aidlab), 2 µl of each primer (10 µM), 2 µl of DNA temple (~30 ng), 34.5 µl ddH2O, and 0.5 µl Red Taq DNA polymerase (5U, Aidlab). PCR was performed using the following procedure: 94 °C for 3 min; followed by 40 cycles of 30 s at 94 °C, annealing for 35 s at 48–56 °C (depending on primer combination), and elongation at 72 °C for 30 s to 4 min (depending on the fragment length); and final extension at 72 °C for 10 min. The PCR products were separated by agarose gel electrophoresis (1% w/v) and purified using a DNA gel extraction kit (Transgen, Beijing, China). The purified products were then ligated into the T-vector (Sangon, Shanghai, China) and sequenced.
Complete Mitogenome Analysis
The graphical map of the complete mitogenome was drawn using the online mitochondrial visualization tool mtviz27. The secondary cloverleaf structure and anticodon of transfer RNAs were identified using the tRNA-scan SE webserver28. Codon usage and the nucleotide composition of the mitogenome were determined using MEGA6. The sequences of 29 Brachyura species and Alpheus distinguendus were aligned using MAFFT29.
Phylogenetic Analysis
Twenty-eight complete Brachyura mitogenomes were downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). In addition, the mitogenome of A. distinguendus was downloaded from GenBank and used as an outgroup taxon. GenBank sequence information is shown in Table 2.
The sequences were aligned with the mitochondrial sequences of closely related species. In order to remove the gaps in sequences, poorly aligned positions and divergent regions were removed using Gblocks25. Then, fasta sequences were converted to nex format sequences and phylip format sequences for Bayesian inference (BI) and Maximum likelihood (ML) analyses using online software (http://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_phylip.php). We used DAMBE to detect the saturation status of the sequences30.
We determined the taxonomic status of C. sinensis within Brachyura by reconstructing the phylogenetic tree. Nucleotide sequences from 30 mitogenome PCGs were combined. The dataset was run using two inference methods: BI and ML analyses. The former was performed using Mrbayes v3.2.131, while ML analysis was performed using raxmlGUI32. The nucleotide substitution model was selected using Akaike information criterion implemented in Mrmodeltest v2.333, 34. The GTR+I+G model was the best model to examine nucleotide phylogenetic analysis and molecular evolution. BI and ML analyses were performed under the GTRCAT model with nucleotide alignment (NT dataset) of the 13 mitochondrial PCGs. ML analyses were performed on 1000 bootstrapped datasets. The BI analysis ran as 4 simultaneous MCMC chains for 10,000,000 generations, sampled every 100 generations, and a burn-in of 5000 generations was used. The average standard deviation of split frequencies was less than 0.01, and the effective sample size determined using tracer v1.6 exceeded 200. These two findings indicate that our data was convergent. The resulting phylogenetic trees were visualized using FigTree v1.4.2.
Results and Discussion
Genome Structure and Organization
The mitogenome of C. sinensis is 15,706 bp long, and its gene content is same as that most known Brachyura: 13 PCGs, 2 rRNA genes, and 22 tRNA genes plus CR (Table 3 and Fig. 1). Twenty-three genes are coded on the J strand and the remaining 14 genes are transcribed on the N strand. It has been deposited in GenBank under accession number KU589292. The genome composition (A: 37.1%, T: 38.6%, C: 14.9%, G: 9.4%) shows a strong A+T bias, which account for 75.7% of the bases, and exhibits a negative AT skew ([A − T]/[A+T] = −0.020) and GC skew ([G − C]/[G+C] = −0.228). The A+T skew of other previously sequenced Brachyura mitogenomes ranged from −0.080 (Pachygrapsus crassipes) to 0.040 (Homologenus malayensis), while the G+C skew ranged from −0.341 (Austinograea rodriguezensis, Geothelphusa dehaani) to −0.219 (Portunus pelagicus) (Table 4). However, different regions have different A+T contents. The CR had the highest A+T content (82.9%), whereas the PCG region had the lowest A+T content (74.2%) (Table 5).

Graphical map of the mitogenome of Clistocoeloma sinensis. Protein-coding and ribosomal RNA genes are shown using standard abbreviations. Genes for transfer RNAs are abbreviated using a single letter. S1 = AGN, S2 = UCN, L1 = CUN, L2 = UUR. CR = control region. The 13 protein-coding genes are yellow, tRNAs are green, rRNAs are red, and CRs are dark red.
Protein-Coding Genes
Among the 13 PCGs, 9 (nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, and cob) were coded on the J strand, while the rest (nad5, nad4, nad4L, and nad1) were on the N strand. The 13 PCGs ranged in size from 159 to 1731 bp (Table 3). Their A+T content was 74.2% and AT skew was −0.026 (Table 5). The relative synonymous codon usage for C. sinensis at the third position is shown in Fig. 2. The usage of both two- and four-fold degenerate codons was biased toward the use of codons abundant in A or T (Table 6), which is consistent with other Brachyura species35,36,37.

Relative synonymous codon usage in Clistocoeloma sinensis mtDNAs. Codon families are provided on the x axis (A). (B) Nucleotide composition conditions.
Transfer RNAs, Ribosomal RNAs, and A+T-Rich Region
Like most Brachyura mtDNA, the C. sinensis mitogenome contains a set of 22 tRNAs genes (Fig. 3), although this feature is not very well conserved in animal mtDNA. The tRNAs ranged in size from 64 to 73 bp and showed a strong A+T bias, as these bases accounted for 76.2% of the DNA. Further, they exhibited a negative AT skew (−0.010) (Table 5). Fourteen tRNA genes were present on the J strand and eight were on the N strand. All the tRNA genes had the typical cloverleaf structure, except for the trnS1 gene, whose dihydroxyuridine arm was instead just a simple loop (Fig. 3). These features are common in most Brachyura mitogenomes35,36,37. The secondary cloverleaf structure of 18 tRNAs was examined using tRNA-scan SE; 4 tRNAs not detected by tRNAscan-SE were found in the unannotated regions by sequence similarity to the tRNAs of other crabs. The 2 rRNA genes with 80.2% total A+T content and positive AT skew (0.007) (Table 5) were located between trnL1 and trnV and between trnV and CR. rrnL is 1336 bp while rrnS is 832 bp. The CR located between rrnS and trnQ, spans 684 bp. This region contains 82.9% AT nucleotides, with a positive AT skew (0.047) and negative GC skew (−0.228) (Table 5).

Secondary structures of the 22 transfer RNA genes of Clistocoeloma sinensis. The tRNAs are labelled with the abbreviations of their corresponding amino acids. Dashes (−) indicate Watson-Crick pairing.
Gene Arrangement
Gene order within the complete mitogenome of C. sinensis is similar to the pancrustacean ground pattern38,39,40 (Fig. 4A), except for the translocation of trnH. Typically, the trnH gene is located between the nad4 and nad5 genes in the pancrustacean ground pattern, but in C. sinensis, it is between the trnE and trnF genes (Fig. 4B). This translocation was also observed in the mitogenomes of Brachyura crabs available in GenBank that were compared with the C. sinensis mitogenome. In addition, in the pancrustacean ground pattern, the tRNA gene order between the CR and nad2 is trnI-trnQ-trnM. However, in C. sinensis, it is trnQ-trnI-trnM (Fig. 4B). The tRNA rearrangements are generally considered to be a consequence of tandem duplication of part of the mitogenome41. Similar non-coding sequences are present at the position of trnI originally occupied by the transposed trnQ in C. sinensis. Because these intergenic sequences have similar lengths to those of typical tRNA genes, they were presumed to be remnants of the trnQ gene and its boundary sequences42. The gene order of C. sinensis is identical to that of S. sinensis (Fig. 4B), which indicates that C. sinensis may belong to the group Sesarmidae of the superfamily Grapsoidea and that C. sinensis and S. sinensis probably belong to sister groups.

Linear representation of gene rearrangements of Brachyura mitogenomes. All genes are transcribed from left to right. tRNA genes are represented by the corresponding single-letter amino acid code. S1 = AGN, S2 = UCN, L1 = CUN, L2 = UUR. CR = control region. rrnL and rrnS are the large and small ribosomal RNA subunits.
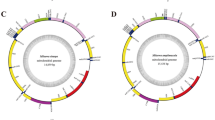
The gene sequences of Varunidae species (Eriocheir japonica sinensis, E. j. hepuensis, E. j. japonica, and Helice latimera) are identical (Fig. 4C). As shown in Fig. 4D, the order and orientation of genes in 7 families are uniform. The order of genes in C. sinensis sequences is different from that in the sequences of the mitogenomes of these 7 families because of the rearrangement of two tRNA genes between CR and trnM: the placement of genes between CR and trnM in C. sinensis is CR-trnQ-trnI-trnM, while that in the 7 families is CR-trnI-trnQ-trnM. In this case, tandem duplication of gene regions may be the most likely mechanism for mitochondrial gene rearrangement, which includes trnI and trnQ, followed by loss of supernumerary genes43, 44. Slipped-strand mispairing occurred first, followed by gene deletion45. Partial PCGs, tRNAs, and rRNAs of Damithrax spinosissimus, G. dehaani, and Xenograpsus testudinatus appear to be rearranged compared to C. sinensis (Fig. 4E–G).
Phylogenetic analysis
Our analyses were based on the NT dataset in mitogenomes derived from 29 Brachyura species belonging to 12 families (Varunidae, Xenograpsidae, Homolidae, Menippidae, Mithracidae, Potamidae, Portunidae, Raninidae, Bythograeidae, Sesarmidae, Grapsidae, and Dotillidae). The data matrix (15,706 bp in all) was analysed using the model-based evolutionary methods of BI and ML analyses (Fig. 5). The ML and BI analyses of the dataset gave the same tree topology. It is obvious that C. sinensis and S. sinensis clustered in one branch in the phylogenetic tree with high nodal support values (Fig. 5), indicating that C. sinensis and S. sinensis have a sister group relationship. This result supported that C. sinensis belongs to Grapsoidea, Sesarmidae. From the phylogenetic tree, we found that X. testudinatus and two Sesarmidae species formed a group and showed close relationships. X. testudinatus, which was originally placed in Varunidae, has been transferred to its own family (Xenograpsidae)21, 46. Analysis of the nucleotide sequences of the 13 mitochondrial PCGs using BI and ML showed that E. j. sinensis, E. j. hepuensis, E. j. japonica, and H. latimera clustered together with high statistical support, showing that these species have a sister group relationship and belong to Grapsoidea, Varunidae. Our phylogenetic analysis indicated that Sesarmidae species, Xenograpsidae species and Varunidae species have close relationships47. In addition, P. crassipes belongs to Grapsoidea, Grapsidae48.

Inferred phylogenetic relationships among Brachyura based on nucleotide sequence of 13 mitochondrial PCGs using maximum likelihood (ML) and Bayesian inference (BI). Alpheus distinguendus was used as the outgroup. The bootstrap value (BP) and Bayesian posterior probability (BPP) of each node are shown as BP based on the NT dataset/BPP based on the NT dataset, 100/1.00.
The phylogenetic position of Ilyoplax deschampsi is always within Grapsoidea21, 47, 49, 50. I. deschampsi belongs to the family Dotillidae, Ocypodoidea. The real phylogenetic position of I. deschampsi should be closer to the Grapsoidea species that shown in Fig. 5. Recent studies on the genus Ucides have also shown similar classification51, 52. G. dehaani belongs to Potamidae, Potamoidea53. However, the phylogenetic tree showed that Potamidae are associated closely with Varunidae, Grapsidae, Sesarmidae, Dotillidae, and Xenograpsidae. This result is in agreement to that inferred from 23 Brachyuran crabs, in which the author use the two mitogenomes21. Phylogenetic relationships between I. deschampsi, G. dehaani and Grapsoidea species need to be reconsidered by integrating more mitogenomic data. More mitogenomic data will also lead to a better overall understanding the phylogenetic relationships among Brachyuran crabs.
Availability of data and materials
The data set supporting the results of this article is available at NCBI (KU589292).
References
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Simon, C. & Frati, F. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Curole, J. P. & Kocher, T. D. Mitogenomics: digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 14, 394–398 (1999).
Lin, C. P. & Danforth, B. N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogen. Evol. 30, 686–702 (2004).
Yu, J. N., Azuma, N. & Abe, S. Genetic differentiation between collections of hatchery and wild masu salmon (Oncorhynchus masou) inferred from mitochondrial and microsatellite DNA analysis. Environ. Biol. Fish. 94, 259–271 (2012).
Liu, Q. N. et al. The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci. Rep. 6, 35878 (2016).
Zhang, S. et al. The application of mitochondrial DNA in phylogeny reconstruction and species identification of portunid crab. Mar. Sci. 32, 9–18 (2008).
Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 110, 15758–15763 (2013).
Zhang, P. et al. Efficient sequencing of Anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol. Biol. Evol. 30, 1899–1915 (2013).
Roe, A. D. & Sperling, F. A. H. Patterns of evolution of mitochondrial cytochrome c oxidase I and II DNA and implications for DNA barcoding. Mol. Phylogenet. Evol. 44, 325–345 (2007).
Foster, P. G. Modeling compositional heterogeneity. Syst. Biol. 53, 485–495 (2004).
Hickson, R. E., Simon, C. & Perrey, S. W. The performance of several multiple-sequence alignment programs in relation to secondary structure features for an rRNA sequence. Mol. Biol. Evol. 17, 530–539 (2000).
Guinot, D., Tavares, M. & Castro, P. Significance of the sexual openings and supplementary structures on the phylogeny of brachyuran crabs (Crustacea, Decapoda, Brachyura), with new nomina for higherranked podotreme taxa. Zootaxa 3665, 1–414 (2013).
Zhou, C., Gong, X., Han, J. & Guo, R. Removal of Pb(II) and Zn(II) from Aqueous Solutions by Raw Crab Shell: A Comparative Study. Water Environ. Res. 88, 374–383 (2016).
Shen, C. J. Description of a new species of crab of the genus Clistocoeloma from China. Ann. Mag. Nat. Hist. 12, 52–59 (1933).
Hsueh, P. W. & Huang, J. F. A new record of Clistocoeloma sinense Shen, 1933, from Taiwan (Decapoda, Brachyura, Grapsidae, Sesarminae), with notes on its distribution and ecology. Crustaceana 69, 63–70 (1996).
Mao, K. Y. Molecular Systematic studies of Thoracotremata Crabs of China and Application of Antennae in the Systematic. East China Normal University (2010)
Sun, H. Y., Zhou, K. Y. & Song, D. X. Mitochondrial genome of the Chinese mitten crab Eriocheir japonica sinenesis (Brachyura: Thoracotremata: Grapsoidea) reveals a novel gene order and two target regions of gene rearrangements. Gene 349, 207–217 (2005).
Tang, B. P., Zhou, K. Y., Song, D. Y., Yang, G. & Dai, A. Y. Molecular systematics of the Asian mitten crabs, genus Eriocheir (Crustacea: Brachyura). Mol. Phylogenet. Evol. 29, 309–316 (2003).
Ji, Y. K., Wang, A. & Lu, X. L. Mitochondrial genomes of two brachyuran crabs (Crustacea: Decapoda) and phylogenetic analysis. J. Crustacean Biol. 34, 494–503 (2014).
Liu, Q. N. et al. The complete mitochondrial genome of Eriocheir japonica sinensis (Decapoda: Varunidae) and its phylogenetic analysis. Biochem. System. Ecol. 62, 241–248 (2015).
Shi, G. H. et al. Unusual sequence features and gene rearrangements of primitive crabs revealed by three complete mitochondrial genomes of Dromiacea. Comp. Biochem. Phys. D 20, 65–73 (2016).
Yang, J. S., Nagasawa, H. & Fujiwara, Y. The complete mitogenome of the hydrothermal vent crab Gandalfus yunohana (Crustacea: Decapoda: Brachyura): a link between the Bythograeoidea and Xanthoidea. Zool. Scr 39, 621–630 (2010).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Abele, L. G. Comparison of morphological and molecular phylogeny of the Decapoda. Mem. Queensl. Mus. 31, 101–108 (1991).
Sun, X. J., Li, Q. & Kong, L. F. Comparative mitochondrial genomics within sea cucumber (Apostichopus japonicus): Provide new insights into relationships among color variants. Aquaculture 309, 280–285 (2010).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Xia, X. & Xie, Z. DAMBE: software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373 (2001).
Ronquist, F., Huelsenbeck, J. P. & Teslenko, M. Draft MrBayes version 3.2 Manual. Tutorials and model summaries. Distributed with the software from http://brahms.biology.rochester.edu/software.html (2011).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012).
Cui, B. K., Tang, L. P. & Dai, Y. C. Morphological and molecular evidences for a new species of Lignosus (Polyporales, Basidiomycota) from tropical China. Mycol. Prog. 10, 267–271 (2011).
Zhao, C. L. & Cui, B. K. A new species of Perenniporia (Polyporales, Basidiomycota) described from southern China based on morphological and molecular characters. Mycol. Prog. 11, 555–560 (2012).
Ma, H. et al. The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene 519, 120–127 (2013).
Ma, H. et al. First mitochondrial genome for the red crab (Charybdis feriata) with implication of phylogenomics and population genetics. Sci. Rep. 5, 11524 (2015).
Ki, J. S., Dahms, H. U. & Hwang, J. S. The complete mitogenome of the hydrothermal vent crab Xenograpsus testudinatus (Decapoda, Brachyura) and comparison with brachyuran crabs. Comp. Biochem. Physiol. D 4, 290–299 (2009).
Boore, J. L., Lvaroa, D. V. & Brown, W. M. Gene translocation links insects and crustaceans. Nature 392, 667–668 (1998).
Podsiadlowski, L. & Braband, A. The complete mitochondrial genome of the sea spider Nymphon gracile (Arthropoda: Pycnogonida). BMC Genomics 7, 284 (2006).
Shen, X. et al. The complete mitochondrial genomes of two common shrimps (Litopenaeus vannamei and Fenneropenaeus chinensis) and their phylogenomic considerations. Gene 403, 98–109 (2007).
Jühling, F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 40, 2833–2845 (2012).
Gong, Y. J., Shi, B. C., Kang, Z. J., Zhang, F. & Wei, S. J. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol. Biol. Rep. 39, 2893–2900 (2012).
Rawlings, T. A., Collins, T. M. & Bieler, R. Changing identities: tRNA duplication and remolding within animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 100, 15700–15705 (2003).
Shi, W., Gong, L. & Wang, S. Y. Tandem duplication and random loss for mitogenome rearrangement in Symphurus (Teleost: Pleuronectiformes). BMC Genomics 16, 355 (2015).
Stanton, D. J., Daehler, L. L., Moritz, C. C. & Brown, W. M. Sequences with the potential to form stem-and-loop structures are associated with coding-region duplications in animal mitochondrial DNA. Genetics 137, 233–241 (1994).
Ng, N. K., Davie, P. J. F. & Schubart, C. D. Xenograpsidae, a new family of crabs (Crustacea: Brachyura) associated with shallow water hydrothermal vents. Raffles. B. Zool. 16, 233–256 (2007).
Schubart, C. D., Cannicci, S. & Vannini, M. Molecular phylogeny of grapsoid crabs (Decapoda, Brachyura) and allies based on two mitochondrial genes and a proposal for refraining from current superfamily classification. J. Zoological Syst. Evol. Res. 44, 193–199 (2006).
Cassone, B. J. & Boulding, E. G. Genetic structure and phylogeography of the lined shore crab, Pachygrapsus crassipes, along the northeastern and western Pacific coasts. Mar. Biol. 149, 213–226 (2006).
Schubart, C. D., Neigel, J. E. & Felder, D. L. Use of the mitochondrial 16S rRNA gene for phylogenetic and population studies of Crustacea. Crustacean Issues 12, 817–830 (2000).
Kitaura, J., Wada, K. & Nishida, M. Molecular phylogeny of grapsoid and ocypodoid crabs with special reference to the genera Metaplax and Macrophthalmus. J. Crustacean Biol. 22, 682–693 (2002).
Broesing, A. & Tuerkay, M. Gastric teeth of some thoracotreme crabs and their contribution to the brachyuran phylogeny. J. Morphol. 272, 1109–1115 (2011).
Ng, N. K., Guinot, D. & Davie, P. J. F. Systema Brachyurorum: part I. an annotated checklist of extant brachyuran crabs of the world. Raffles. B. Zool. 17, 1–286 (2008).
Okano, T., Suzuki, H. & Miura, T. Comparative biology of two Japanese freshwater crabs Geothelphusa exigua and G. dehaani (Decapoda, Brachyura, Potamidae). J. Crustacean Biol. 20, 299–308 (2000).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31672267 and 31640074), the Natural Science Foundation of Jiangsu Province (BK20160444), the Natural Science Foundation of Education Department of Jiangsu Province (15KJB240002, 12KJA180009, and 16KJA180008), the Jiangsu Provincial Key Laboratory of Coastal Wetland Bioresources and Environmental Protection (JLCBE14006), the Jiangsu Key Laboratory for Bioresources of Saline Soils (JKLBS2014013 and JKLBS2015004), the Yancheng City Agricultural Science and Technology Guiding Project (YKN2014022).
Author information
Authors and Affiliations
Contributions
Q.N.L. and B.P.T. conceived and designed the experiments. Q.N.L., Z.Z.X., and X.Y.C. performed the experiments. Q.N.L., Z.F.W., Y.L., H.B.Z. and Z.Z.X. analyzed the data. D.Z.Z., C.L.Z. and B.P.T. contributed reagents and materials. Q.N.L. and Z.Z.X. wrote the paper. Z.Z.X., and Q.N.L. revised the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xin, ZZ., Liu, Y., Zhang, DZ. et al. Complete mitochondrial genome of Clistocoeloma sinensis (Brachyura: Grapsoidea): Gene rearrangements and higher-level phylogeny of the Brachyura. Sci Rep 7, 4128 (2017). https://doi.org/10.1038/s41598-017-04489-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04489-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
