
Abstract
Base excision repair (BER) is a major pathway for removal of DNA base lesions and maintenance of genomic stability, which is essential in cancer prevention. DNA glycosylases recognize and remove specific lesions in the first step of BER. The existence of a number of these enzymes with overlapping substrate specificities has been thought to be the reason why single knock-out models of individual DNA glycosylases are not cancer prone. In this work we have characterized DNA glycosylases NEIL1 and NEIL2 (Neil1 −/− /Neil2 −/−) double and NEIL1, NEIL2 and NEIL3 (Neil1 −/− /Neil2 −/− /Neil3 −/−) triple knock-out mouse models. Unexpectedly, our results show that these mice are not prone to cancer and have no elevated mutation frequencies under normal physiological conditions. Moreover, telomere length is not affected and there was no accumulation of oxidative DNA damage compared to wild-type mice. These results strengthen the hypothesis that the NEIL enzymes are not simply back-up enzymes for each other but enzymes that have distinct functions beyond canonical repair.
Similar content being viewed by others


Introduction
An inevitable consequence of aerobic metabolism is the production of reactive oxygen species (ROS)1, 2. In addition, exogenous sources such as cigarette smoking, pollutants, excessive alcohol consumption, asbestos, exposure to ionizing radiation, and bacterial, fungal or viral infections lead to enhanced generation of ROS3,4,5,6,7,8. ROS are well-described second messengers in a variety of cellular processes but ROS can be harmful at high concentrations becoming a threat to cells by causing peroxidation of lipids, oxidation of proteins, damage to nucleic acids and enzyme inhibition9,10,11,12,13,14. DNA lesions resulting from this type of damage are mutagenic and cytotoxic and, if not repaired, can cause genetic instability that may lead to various pathological conditions including neurodegenerative and cardiovascular diseases, carcinogenesis and premature aging15,16,17,18. The Base Excision Repair (BER) pathway is the primary mechanism responsible for the removal of small, non-helix-distorting base lesions that arise from oxidation as well as deamination and alkylation19. DNA glycosylases initiate BER by recognizing and excising the modified base, thereby forming an apurinic/apyrimidinic site (AP) site before repair is completed by the action of several enzymes that ultimately restore DNA integrity19.
Six mammalian DNA glycosylases initiate BER of a broad spectrum of oxidative DNA base lesions: E. coli Nei endonuclease VIII-like (NEIL) NEIL1, NEIL2 and NEIL3, 8-oxoguanine DNA glycosylase (OGG1), Escherichia coli (E. coli) MutY homologue (MUTYH) and E. coli Nth endonuclease III-like 1 (NTHL1)19, 20. 8-oxo-7,8-dihydroguanine (8-oxoG) is a major DNA oxidized lesion removed by OGG121, 22. Further oxidation of 8-oxoG results in the formation of spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh) that can both mispair with adenine and guanine23,24,25,26. The NEIL proteins have been shown to recognize and excise a number of oxidized bases including both Sp and Gh, from different substrates such as double- and single-stranded DNA, bubble DNA structures and telomeric and promoter quadruplex DNA structures27,28,29,30,31,32,33.
Thousands of endogenous DNA lesions are generated per day per cell making BER an essential pathway in preventing genomic damage and instability as seen in the numerous phenotypes observed in BER-deficient models. While deficiency of the common downstream enzymes of BER and the thymine-DNA glycosylase (TDG/Tdg) are embryonic lethal, the remaining DNA glycosylase-deficient mouse models have failed to display elevated mutation rates, increased cancer incidence or other severe phenotypes in the absence of any mutagenic insult34,35,36,37,38,39,40. This lack of altered phenotype exhibited by the single DNA glycosylase-deficient models has been ascribed to redundancy, or so-called back-up activities, between the enzymes as well as repair by alternative repair pathways.
In this paper, we describe the generation of single NEIL1 and NEIL2 (Neil1 −/− and Neil2 −/−), NEIL1/NEIL2 double (Neil1 −/− /Neil2 −/−) and NEIL1/NEIL2/NEIL3 triple (Neil1 −/− /Neil2 −/− /Neil3 −/−) knock-out mouse models. We have used these models to study spontaneous tumour formation. Furthermore, the double and triple knock-out models have been used to determine oxidative damage accumulation in genomic DNA, relative telomere length and mutation frequencies in peripheral blood erythrocytes using the in vivo Pig-a gene mutation assay. Finally, we have derived primary mouse embryonic fibroblasts (MEFs) from our mouse models and determined cell survival after exposure to potassium dichromate.
Results and Discussion
Generation and characterization of Neil1 −/−, Neil2 −/− , Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− mice
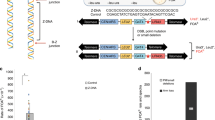
Conditional NEIL1 knock-out mice were generated by using the Cre-loxP recombination system. We made a gene-targeting vector in which the loxP sites flanked exon 2 of Neil1 and a positive selection cassette for neomycin resistance flanked by FRT sequences (Fig. 1a and b). Exon 2 of NEIL1 contains the N terminal catalytic proline that forms a Schiff base with the oxidized lesion41, 42. The targeting construct was linearized with NdeI and electroporated into 129/SvJ embryonic stem cells (ESCs) to obtain the recombined Neil1 locus (Fig. 1c). Genomic DNA was extracted from Geneticin-resistant ESC clones, NdeI-digested, and subjected to Southern blot analysis (Fig. 1d). One positive clone was identified (G4) and correct 5′ and 3′ targeting was confirmed by PCR. Recombinant ESCs derived from the G4 clone were injected into C57BL/6 blastocysts, which were transferred to pseudo-pregnant foster mothers (Norwegian Transgenic Center, Institute of Basic Medical Science, Faculty of Medicine, University of Oslo, Oslo, Norway). Chimeric male offspring were bred with C57BL/6N females to obtain Neil1 +/fl ° xne ° mice. Cross-breeding with FLP recombinase-expressing mice was performed to remove the neomycin cassette (Fig. 1e). Cross-breeding with CMV-Cre-expressing mice was performed to remove the floxed exon 2 in all tissues (Fig. 1f). Inter-crossing of F1 Neil1 +/− mice resulted in offspring of the expected Mendelian ratios. We examined Neil1 expression levels by RT-PCR in brain tissue of Neil1 +/+ and Neil1 −/− mice and as expected Neil1 transcripts were absent in the knock-out mice (Fig. 1g). As previously observed by Vartanian and colleagues, Neil1 −/− mice were viable and developed obesity late in life36.

Targeted disruption of the murine Neil1 locus by conditional knockout strategy. (a) The Neil1 locus from the 129/SvJ mouse genome is shown. Exons, NdeI restriction enzyme sites, the position of the 5′ and 3′ probes used for Southern blotting and different primers used for genotyping during the generation of the Neil1 −/− mice, are indicated. (b) The targeting construct carried two homology arms (2 kb and 6.2 kb), floxed exon 2 (Ex2) and an FRT-flanked neo cassette (NEO). (c) The targeted Neil1 locus is shown. The positions of the 5′ and 3′ Southern blot probes are indicated. B and c The loxP sites are indicated by white triangles and the Frt sites are indicated by black triangles. Some restriction enzyme sites are shown; N: NdeI, E: EcoRI, B: BamHI, Bg: BglII, X: XhoI. (d) Southern blot analysis of DNA samples from two Geneticin-resistant ES clones digested with NdeI and hybridized with the 5′ probe identified a positive clone (G4). (e) Typical genotyping results obtained by PCR after breeding Neil1 +/floxneo mice with FLP recombinase expressing mice to remove the FRT-flanked neo marker. (f) Typical genotyping results obtained by PCR after breeding Neil1 +/flox mice with Cre recombinase expressing mice to remove the floxed exon 2 to generate Nei1l −/− mice. (g) Disruption of Neil1 expression in Nei1l −/− mice was measured by RT-PCR analysis of mRNA isolated from brain. 1 and 2: WT; 3 and 4: Nei1l −/−. (e–g) M: GeneRuler DNA ladder mix.
Neil2 +/− mice were purchased from the Texas A&M Institute for Genomic Medicine, TX, USA. Briefly, Neil2 +/− mice were generated using a 129/SvEv ES cell clone (Omnibank Clone OST218883) that contained a gene-trap vector insertion (Gene trapping vector VICTR48) within the first intron of Neil2. The insertion produced incorrect splicing, perturbing the expression of all Neil2 downstream exons (Fig. 2a and b). Neil2 −/− mice were generated by inter-crossing of F1 Neil2 +/− mice. A PCR-based genotyping assay was established to distinguish wild-type and homozygous mice (Fig. 2c). Inter-crossing of Neil2 +/− mice resulted in offspring of the expected Mendelian ratios. Neil2 transcript levels were determined by RT-PCR in brain and thymus of Nei2 +/+ and Neil2 −/− mice. No transcripts were detected in any of the organs tested (Fig. 2d). As observed by Chakraborty and co-workers40, the mice were viable and fertile and did not display any overt phenotype.

Targeted disruption of the murine Neil2 locus by gene trapping. (a) The Neil2 locus from the 129/SvJ mouse genome is shown with indicated exons. A retroviral gene trap vector was used to produce a clone: OST218883, which contains an insertion within the first intron of Neil2. This clone was used to generate Neil2 −/− mice. (b) Detail of the Neil2 locus from the 129/SvJ mouse genome and targeted Neil2 locus showing the position of the primers used for genotyping. (c) Typical genotyping results obtained by PCR after breeding Neil2 +/− mice with Neil2 +/− mice. (d) Disruption of Nei12 expression in Neil2 −/− mice was measured by RT-PCR analysis of mRNA isolated from brain and thymus. 1: WT brain, 2: WT thymus, 3: Neil2 −/− brain and 4: Nei12 −/− thymus. (c and d) M: GeneRuler DNA ladder mix.
Neil1 −/− /Neil2 −/− mice were generated by cross-breeding Neil1 −/+ /Neil2 +/+ mice with Neil1 +/+ /Neil2 −/+ mice (Supplementary Table 1). The resulting Neil1 +/− /Neil2 +/− offspring was inter-crossed and genotyped to identify Neil1 −/− /Neil2 −/− mice (Supplementary Fig. 1). Neil1 −/− /Neil2 −/− /Neil3 −/− mice were generated by crossing Neil1 −/− /Neil2 +/− /Neil3 +/+ mice with Neil1 +/+ /Neil2 +/+ /Neil3 −/− mice, generated previously in our lab37 (Supplementary Table 2). Offspring was cross-bred and genotyped until Neil1 −/− /Neil2 −/− /Neil3 −/− mice were identified (Supplementary Fig. 1). Offspring of Neil1 −/− /Neil2 −/− × Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− × Neil1 −/− /Neil2 −/− /Neil3 −/− breedings displayed no viability issues as indicated by normal weight gain and normal litter sizes as compared to the wild-type and single knock-out counterparts. The obese phenotype observed in Neil1 −/− mice disappeared in the double and triple knock-out models. As for the single knock-outs, the double and triple knock-outs did not display any overt phenotype.
We looked for signs of stress such as loss of appetite and weight loss, lethargy, rough hair coat in colonies of wild-type, single, double and triple knock-outs. Mice that presented any of these signs were promptly euthanized and subjected to necropsy. We observed sporadic tumours in all strains including wild-type animals, however there was no overall difference in tumour formation between any of the knock-out models compared to the wild-type mice. Thus, none of the mouse models presented in this paper displayed increase in spontaneous tumour formation within 20 months. In previous DNA glycosylase-deficient mouse models such as OGG1 and MUTHY, the genetic background appeared to have an effect on the susceptibility to tumour formation43,44,45,46,47. However, our results are in agreement with previous studies of single NEIL1 and NEIL2 knock-out mice carried out in several laboratories34, 36, 37, 40.
Mice deficient in DNA glycosylases involved in removing oxidative bases were firstly developed to reveal the role of BER in tumorigenesis. Surprisingly, both NEIL1 and OGG1 deficient mice have been reported to develop late-onset obesity36, 48, 49. However, not all colonies of Neil1 −/− or Ogg1 −/− mice have displayed such a phenotype50, 51. Our Neil1 −/− mice presented an obese phenotype that disappeared in the double and triple knock-out mice. At present we do not know the reason but due to the variability in penetrance of the phenotype, studies to address this would take many years and thousands of mice48. Some double knock-out mouse models like Ogg1 −/− /Mutyh −/− and Nthl1 −/− /Neil1 −/− are predisposed to develop cancer, while others such as Ogg1 −/− /Nthl1 −/− are not46, 51, 52. Interestingly, neither the double nor the triple knock-out models presented in this work displayed any sign of augmented spontaneous tumour formation. Accordingly, NTHL1 seems to play a role in removal of mutagenic pyrimidine lesions, while the nucleotide incision repair (NIR) can constitute repair activity against a subset of oxidized pyrimidines53, 54. Furthermore, repair activity towards hydantoins by the nucleotide excision repair (NER) in mammalian cells was recently reported55. Altogether, cells appear to be robustly equipped with a battery of mechanisms, to withstand normal physiological oxidative stress that could explain the lack of phenotype in our DNA glycosylase-deficient models.
The mice generated in the present work can now be crossed with other NER-deficient mouse models to assess the consequences of hydantoin accumulation and to evaluate the contribution of the different pathways to the repair of hydantoins in different tissues. Furthermore, the mice can be exposed to different oxidative insults to assess tumour formation.
Normal telomere lengths in P0 and two year-old kidney, liver and spleen tissue
Recent work has shown the likely involvement of the NEILs in both telomere maintenance and gene regulation32, 33, 40. Furthermore, telomere dysfunction has been shown to occur in NTHL1, OGG1, NEIL2 and UNG-deficient mouse models40, 56,57,58. We thus investigated spontaneous telomere length variation in liver, kidney and spleen from new-born and two year-old Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− animals. As illustrated in Fig. 3 there was no significant change in the average telomere length in the double or triple knock-outs compared to wild-type in tissue harvested from two year-old mice. The same was true for tissue derived from new-born mice (data not shown).

Quantitative analysis of telomere length. Relative telomere length was assessed in kidney, spleen and liver tissue isolated from three individual animals using qPCR in 2 year-old Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− tissue compared to WT. Telomere length (mean ± SD) was presented relative to a single copy gene (36B4). Neil1/2 −/−: Neil1 −/− /Neil2 −/−; Neil1/2/3 −/−: Neil1 −/− /Neil2 −/− /Neil3 −/−.
Organs from two year-old NEIL enzymes-deficient mice showed no increase in overall steady state levels of oxidized DNA base lesions
The NEIL proteins catalyze the excision of a broad range of oxidized bases27,28,29,30,31,32,33. We measured the levels of 8-oxoG and 5-OHC in genomic DNA derived from tissue isolated from two year-old animals. 5-OHC is a substrate for all the three NEIL enzymes whereas 8-oxoG is a good marker to indicate the extent of cellular oxidative stress. As seen in Fig. 4, the steady state levels of 8-oxoG and 5-OHC, in liver, kidney and spleen, did not differ between the two genotypes compared to wild-type. Sp and Gh, the secondary oxidation products of 8-oxoG have been detected both in E. coli and in murine liver and colon59, 60. However, the levels reported were nearly 100 times lower than 8-oxoG. We have tried to measure the in vivo levels of Sp and Gh in several tissues from our NEILs-deficient mice but failed so far. Our protocols are not sensitive enough.

No accumulation of oxidized DNA bases. Oxidized DNA bases were measured in 2 year-old WT, Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− tissue isolated from three individual animals. LC-MS/MS quantification of (a) 8-oxoG and (b) 5-OHC in kidney, spleen and liver. Data are presented as mean (±SD). Neil1/2 −/−: Neil1 −/− /Neil2 −/−; Neil1/2/3 −/−: Neil1 −/− /Neil2 −/− /Neil3 −/−. dNs: deoxyribonucleotides.
Spontaneous phenotypic mutation frequencies in Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− mice measured at the Pig-a gene
To elucidate whether the double or triple knock-out mouse models were associated with increased spontaneous phenotypic mutation frequencies we used the Pig-a gene mutation assay61,62,63,64,65. This assay is based on a cell surface phenotype characterized by the absence of membrane glycosylphosphatidylinositol (GPI)-anchored proteins such as CD24, CD48, CD59 and CD5566. Negative mutant hematopoietic cells were identified by fluorescence-activated cell sorting analysis. We measured mutation frequencies based on loss of a surface marker CD24 in immature RETs and mature RBCs. A definite advantage of the Pig-a gene mutation assay when compared to well-established mutagenicity tests like the HPRT or TK/MLA assay is the comparatively easy and fast procedure that interrogates high numbers of potential mutant cells (3–100 × 106 cells/sample).
Mammals posses three enzymes that prevent mutagenesis caused by 8-oxoG. OGG1 excises 8-oxoG from DNA, MUTYH removes adenine misincorporated opposite 8-oxoG in DNA and MTH1 (mutT homologue 1, NUDT1) degrades 8-oxodGTP in the nucleotide pool to prevent its incorporation into DNA21, 22, 67, 68. Since spontaneous and induced mutation frequencies in the single Ogg1 −/−and Muthy −/− and the double Ogg1 −/− /Mutyh −/− mouse models have been reported, we tested the sensitivity of the Pig-a gene mutation assay using 6 months-old single Muthy −/− and Ogg1 −/− and double Ogg1 −/− /Mutyh −/− mice43, 44, 46, 69,70,71. Increased mutagenic frequencies were detected in both RETs and in RBCs in the Ogg1 −/− /Mutyh −/− double knock-out mice compared to the wild-type mice, but not in the single Ogg1 −/−or Muthy −/− knock-out mice (Fig. 5c). To exclude the possibility that bone marrow cytotoxicity was the cause of the observed increase of spontaneous mutation frequencies, the percentage of RETs (% RET, ratio of newly formed RNA-positive erythrocytes relative to all erythrocytes) was calculated. As presented in Fig. 5c, lower panel, there was no significant change in the relative number of RETS in any of the mutants. Neither the 18 months-old Neil1 −/− /Neil2 −/− mice nor the 23 months-old Neil1 −/− /Neil2 −/− /Neil3 −/− mice displayed significantly increased Pig-a gene mutation frequencies (Fig. 5a and b).

Phenotypic mutation frequencies measured by the Pig-a gene mutation assay. Frequencies of mutant phenotype reticulocytes (RETCD24−), erythrocytes (RBCCD24−) and percentage reticulocytes (% RET) were determined in (a) 18 months-old WT (n = 8) and Neil1 −/− /Neil2 −/− (Neil1/2 −/−) (n = 13); (b) 23 months-old WT (n = 2) and Neil1 −/− /Neil2 −/− /Neil3 −/− (Neil1/2/3 −/−) (n = 17) mice and c) 6 months-old WT (n = 3), Mutyh −/− (M −/−) (n = 3), Ogg1 −/− /Mutyh −/− (O/M −/−) (n = 3), and Ogg1 −/− (O −/− ) (n = 4) mice. Results are presented for three separate experiments (a–c). Each data point represents one mouse; and groups are summarized as box-plots of at least two individuals. Groups that are significant different (p < 0.05) from control group (WT) are marked with an asterisk dependent on the distribution of the data, that is (a) non-parametric Wilcoxon test, (b) t-test and (c) Dunnett’s test.
It can be argued that the Pig-a gene mutation assay was not the best method to measure spontaneous mutation frequencies. However, its sensitivity is comparable to that of the Hprt assay and an increase in phenotypic mutation frequencies was detected in the Ogg1 −/− /Mutyh −/− double knock-out mice72. Altogether, the results presented endorse the Pig-a gene mutation assay as an in vivo mutation test that is sensitive, fast, reliable and less costly than other tests.
Neither hydrogen peroxide nor potassium dichromate treatment affect the survival of NEILs-deficient MEF cells
Because we did not observe any phenotype under normal conditions, we wanted to investigate if exposure of MEFs to DNA damaging agents such as hydrogen peroxide or potassium dichromate had any consequences. Further oxidation of 8-oxoG by for example high valent metals such as chromium (V) results in the generation of the lesions Sp and Gh. In E. coli, these lesions are repaired by both Fpg and Nei and in mammalian cells by the NEIL proteins and NTHL1. Moreover, it has been shown that Nei-deficient E. coli are sensitive to chromate exposure59. Cells were exposed to increasing amounts of potassium dichromate and hydrogen peroxide for 24 hours and cell viability was measured using the MTT assay. As shown in Fig. 6a none of the DNA glycosylase-deficient MEFs were more sensitive to potassium dichromate than wild-type MEF cells. We observed significant sensitivity of Neil1 −/− /Neil2 −/− MEFs compared to wild-type towards hydrogen peroxide at only one concentration (Fig. 6b). However, none of the other MEFs were significantly more sensitive compared to wild-type cells. A narrow window of vulnerability cannot be ruled out, but in general, NEIL1, NEIL2 and/or NEIL3 deficiency is not enough to render MEF cells sensitive towards hydrogen peroxide or potassium dichromate. This lack of sensitivity is most likely due to the removal of oxidized lesions including Sp and Gh by other DNA glycosylases such as NTHL1 and by other pathways. NER and DNA mismatch repair (MMR) have also been shown to have a role in the repair of oxidative DNA damage73, 74. More recently, NER has been shown to mediate repair of hydantoin lesions in cells55.

Primary MEFs survival response to hydrogen peroxide and potassium dichromate. Cell survival response towards increasing concentrations of (a) potassium dichromate and (b) hydrogen peroxide was measured using the MTT assay after 24 hour exposure. MEF cells: N1 −/−: Neil1 −/−, N2 −/−: Neil2 −/−: N3 −/−: Neil3 −/−, Neil1/2 −/−: Neil1 −/− /Neil2 −/−, Neil1/2/3 −/−: Neil1 −/− /Neil2 −/− /Neil3 −/−. Results are mean (±SD) OD (% of control) of three to five independent experiments for each genotype. *p < 0.005 compared to WT.
Conclusions
The results presented in this paper strongly indicate that the NEIL DNA glycosylases are not back-up enzymes for each other in processing of oxidized DNA bases. NEIL1 has been implicated in the repair of base lesions during replication75,76,77, whereas NEIL2 has been suggested to be relevant for transcription associated BER40, 78. NEIL3-dependent repair of oxidative DNA damage appears to be important in proliferating cells, including neural stem/progenitor cells required for induced and continuous neurogenesis37, 79. Altogether it is plausible to suggest that NEIL1, NEIL2 and NEIL3 DNA glycosylases have evolved to carry out different specialized functions that do not overlap. We hypothesize that the enzymes carry out specific functions that are coordinated or associated with cellular processes such as cell cycle progression, transcription, replication and chromatin remodelling.
Methods
Ethics statement
This study was carried out in accordance with the Norwegian Regulation on Animal Experimentation, which is based upon the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes. All experiments were approved by the Norwegian Animal Research Committee.
Reagents
Lympholyte®-Mammal cell separation reagent was purchased from CedarLane (Burlington, ON, Canada). Anti-PE MicroBeads, LS + Positive Selection Columns and a QuadroMACS™ Separator were from Miltenyi Biotec (GmbH, Bergisch Gladbach, Germany). CountBright™ Absolute Counting Beads were from Invitrogen, Life Technologies™ (Carlsbad, CA, USA). Heat-inactivated fetal bovine serum (FCS) was from PAA Laboratories (Pasching, Austria). DMEM medium, trypsin/EDTA solution, penicillin/streptomycin was from BioWhittaker™ (Walkersville, MD, USA). GlutaMAX™ Supplement, was from Gibco (Grand Island, NY, USA). Anticoagulant solution, buffered salt solution, nucleic acid dye solution (SYTO®13), anti-CD24-PE and anti-CD61-PE were from the Prototype In Vivo Mouse MutaFlow® Kit, Litron Laboratories (Rochester, NY, USA). N-ethyl-N-nitrosourea (ENU, CAS 759–73–9, cat. no. N3385), potassium dichromate (cat. no. 207802), Hydrogen peroxide solution 30 wt. % in H2O (cat. no. 216763) and DMSO were from Sigma Aldrich (St. Louis, MO, USA). Cell proliferation kit (MTT) (cat. no. 11465007001) was from Roche Diagnostic (Roche Inc., Mannheim, Germany).
Primers
Primers used for genotyping and RT-PCR are listed in Table 1.
Animals
All strains were back-crossed more than 8 generations onto a C57BL/6N background (N8 = 99.6%). A total of 55 animals were used for the Pig-a gene mutation experiments: 3 wild-type (WT), 3 Mutyh −/−, 4 Ogg1 −/− and 3 Ogg1 −/− /Mutyh −/− 6 months-old; 8 WT and 13 Neil1 −/− /Neil2 −/− 18 months-old and 2 WT and 17 Neil1 −/− /Neil2 −/− /Neil3 −/− 23 months-old mice. For a positive control of the Pig-a gene mutation assay, 2 WT mice were treated with ENU (i.p. injection, 40 mg/kg bodyweight (bw) on three consecutive days, total dose of 120 mg/kg bw).
Longevity studies
Mice were allowed to age and were observed for development of disease and subject to full necropsy at age between 18 and 24 months-old.
Primary mouse embryonic fibroblasts and exposure experiments
Primary mouse embryonic fibroblasts (MEFs) were generated from E14.5-E-15.5 mouse tissue of WT, Neil1 −/−, Neil2 −/−, Neil3 −/−, Neil1 −/− /Neil2 −/− and Neil1 −/− /Neil2 −/− /Neil3 −/− embryos. Limbs were removed from embryos, the tissue was chopped into small pieces and cell suspension was made using a pipette. MEFs were grown in DMEM supplemented with 10% serum, 2 mM glutamine and penicillin/streptomycin. Cells grown for 3–4 days were trypsinized and frozen in complete medium with 10% DMSO as stock. After thawing, cells were grown in complete medium for 2–4 days before using them for survival assays. Primary MEFs were seeded in 96 well plates at a concentration of either 4000 or 8000 cells/well, incubated for 4 h and then treated with increasing amounts of potassium dichromate (K2Cr2O7) or hydrogen peroxide (H2O2) for 24 h. Results were calculated from at least three independent experiments carried out in quadruplicate. Cell viability was determined with the MTT Cell Proliferation kit (Roche Diagnostics) and absorbance was measured on a Wallac 1420 Multilabel Counter at a wavelength of 570 nm and reference wavelength 670 nm. Cell viability was calculated as percentage relative to control cells set as 100%.
Mutation frequencies at the Pig-a gene
Blood samples were taken from the saphenous vein using a 21-G needle and heparinised capillary tube. 60 µl of free-flowing blood were added to 100 µl anticoagulant, mixed well and kept at room temperature until further processing within two hours. The Pig-a gene mutation assay was performed as described previously61,62,63 and in the Prototype Mouse MutaFlow® Kit instruction manual (version120301). At least 100 × 106 red blood cells (RBC) and 3 × 106 reticulocytes (RET) per sample were recorded. The Prototype MutaFlow® protocol used herein included a post-column incubation step of 30 min at 37 °C, a step that was discovered to potentially lyse mouse erythrocytes, especially RNA-negative erythrocytes. This implies that the RBCCD24− values presented herein may be somewhat underestimated.
The data for Pig-a gene mutation assay was processed and calculated as described in ref. 80 using Microsoft Excel 2010. Data was log(10) transformed (an offset of 0.1 was added for RETCD24− and RBCCD24− values prior transformation to avoid zero) and further statistics was performed depending on the distribution using JMP Pro 12 (Statistical Analysis System Institute Inc., Cary, NC, USA). That is, t-test for data from 23 months-old Neil1 −/− /Neil2 −/− /Neil3 −/− mice; non-parametric Wilcoxon test for 18 months-old Neil1 −/− /Neil2 −/− mice and Dunnett’s for 6 months-old Ogg1 −/− /Mutyh −/− mice.
DNA extraction
DNA was isolated from P0 and two year-old tissues using DNeasy Blood and Tissue Kit (Qiagen, 69506), according to the manufacturer’s protocol.
Telomere relative length quantification
Relative telomere length was determined by qPCR as described81. Briefly, after quantifying genomic DNA extracted from each sample, real-time SYBR Green qPCR was performed on a Step One Plus Real-Time PCR system (Applied Biosystems) using two pairs of primers, one telomere specific and one single copy gene (36B4). Relative telomere relative length quantification was determined using the relative standard curve method. Each sample was loaded in triplicate.
LC-MS/MS analysis of 8-oxoG and 5-hydroxycytosine in genomic DNA
Two µg of genomic DNA was enzymatically hydrolyzed to deoxyribonucleosides by incubation in a mixture of benzonase (sc-391121B, Santa Cruz Biotechnology, Heidelberg, Germany) nuclease P1 from Penicillium citrinum (N8630, Sigma Aldrich, St. Louis, MO, USA) and alkaline phosphatase from E. coli (P5931, Sigma Aldrich, St. Louis, MO, USA) in 10 mM ammonium acetate buffer, pH 5.3 at 40 °C for 40 min. Three volume equivalents of ice-cold methanol were added to the reactions after digestion was completed to precipitate proteinaceous contaminants. Following centrifugation at 16000 × g at 4 °C for 40 min the supernatants were collected in new tubes and dried under vacuum at room temperature. The resulting residues were dissolved in 50 µl of water for LC-MS/MS quantification of 8-oxoG and 5-hydroxydeoxycytosine (5-OHC). For quantification of unmodified nucleosides (dA, dC, dG, dT) samples were further diluted with water up to 1:5000. Chromatographic separation of nucleosides was performed using a Shimadzu Prominence LC-20AD HPLC system with an Ascentis Express C18 2.7 µm 150 × 2.1 mm i.d. column equipped with an Ascentis Express Cartridge Guard Column (Supelco Analytical, Bellefonte, PA, USA) with EXP Titanium Hybrid Ferrule (Optimize Technologies Inc.) at a flow rate of 0.14 ml/min at ambient temperature. The mobile phase consisted of A (0.1% formic acid in water) and B (0.1% formic acid in methanol). The following conditions were employed during chromatographic separation: unmodified nucleosides –starting with 90% A and 10% B for 0.1 min, followed by a 2.4 min linear gradient of 10–60% B, and 4.5 min re-equilibration with the initial mobile phase conditions; 8-oxoG and 5-OHC – starting with 95% A and 5% B for 0.5 min, followed by a 7.5 min linear gradient of 5–45% B, and 5.5 min re-equilibration with the initial mobile phase conditions. Online mass spectrometry detection was performed using an Applied Biosystems/MDS Sciex API5000 Triple quadrupole mass spectrometer (ABsciex, Toronto, Canada), operating in positive electrospray ionization mode. The deoxyribonucleosides were monitored by multiple reaction monitoring using mass transitions 252.2 → 136.1 (dA), 228.2 → 112.1 (dC), 268.2 → 152.1 (dG), 243.2 → 127.0 (dT), 284.1 → 168.1 (8-oxoG) and 244.1 → 128.0 (5-OHC).
References
Lambeth, J. D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 (2004).
Dröse, S. & Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol 748, 145–169 (2012).
Stehbens, W. E. Oxidative stress in viral hepatitis and AIDS. Exp. Mol. Pathol. 77, 121–132 (2004).
Valko, M., Rhodes, C. J., Moncol, J., Izakovic, M. & Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 160, 1–40 (2006).
Das, S. K. & Vasudevan, D. M. Alcohol-induced oxidative stress. Life Sci. 81, 177–187 (2007).
Yang, W. & Omaye, S. T. Air pollutants, oxidative stress and human health. Mutat. Res. 674, 45–54 (2009).
Jomova, K. et al. Arsenic: toxicity, oxidative stress and human disease. J. Appl. Toxicol. 31, 95–107 (2011).
Chew, S. H. & Toyokuni, S. Malignant mesothelioma as an oxidative stress-induced cancer: An update. Free Radic. Biol. Med. 86, 166–178 (2015).
Uchida, K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 42, 318–343 (2003).
Davies, M. J. The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 (2005).
Hamanaka, R. B. & Chandel, N. S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513 (2010).
Dizdaroglu, M. & Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 46, 382–419 (2012).
Jacob, K. D., Noren Hooten, N., Trzeciak, A. R. & Evans, M. K. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech. Ageing Dev. 134, 139–157 (2013).
Knoefler, D. et al. About the dangers, costs and benefits of living an aerobic lifestyle. Biochem. Soc. Trans. 42, 917–921 (2014).
Chen, X., Guo, C. & Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res 7, 376–385 (2012).
Sohal, R. S. & Orr, W. C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 52, 539–555 (2012).
Elnakish, M. T., Hassanain, H. H., Janssen, P. M., Angelos, M. G. & Khan, M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J. Pathol. 231, 290–300 (2013).
Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res 763, 212–245 (2015).
Krokan, H. E. et al. Base excision repair. Cold Spring Harb. Perspect. Biol 5, a012583, doi:10.1101/cshperspect.a012583 (2013).
Wallace, S. S. DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagen. 54, 691–704 (2013).
Bjorås, M. et al. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J 16, 6314–6322 (1997).
Radicella, J. P., Dherin, C., Desmaze, C., Fox, M. S. & Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 94, 8010–8015 (1997).
Luo, W., Muller, J. G., Rachlin, E. M. & Burrows, C. J. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett. 2, 613–616 (2000).
Luo, W., Muller, J. G., Rachlin, E. M. & Burrows, C. J. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem. Res. Toxicol. 14, 927–938 (2001).
Kornyushyna, O., Berges, A. M., Muller, J. G. & Burrows, C. J. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment). Biochemistry 41, 15304–15314 (2002).
Duarte., V., Muller, J. G. & Burrows, C. J. Insertion of dGMP and dAMP during in vitro DNA synthesis opposite an oxidized form of 7,8-dihydro-8-oxoguanine. Nucleic Acids Res 27, 496–502 (1999).
Morland, I. et al. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res 30, 4926–4936 (2002).
Hazra, T. K. et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 277, 30417–30420 (2002).
Hailer, M. K., Slade, P. G., Martin, B. D., Rosenquist, T. A. & Sugden, K. D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 4, 41–50 (2005).
Liu, M. et al. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 107, 4925–4930 (2010).
Krokeide, S. Z. et al. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair 12, 1159–1164 (2013).
Zhou, J., Fleming, A. M., Averill, A. M., Burrows, C. J. & Wallace, S. S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res 43, 4039–4054 (2015).
Fleming, A. M., Zhou, J., Wallace, S. S. & Burrows, C. J. A role for the rifth G-track in G-quadruplex forming oncogene promoter sequences during oxidative stress: Do these “spare tires” have an evolved function? ACS Cent. Sci 1, 226–233 (2015).
Torisu, K., Tsuchimoto, D., Ohnishi, Y. & Nakabeppu, Y. Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease VIII-like protein. J. Biochem. 138, 763–772 (2005).
Friedberg, E. C. & Meira, L. B. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair 5, 189–209 (2006).
Vartanian, V. et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 103, 1864–1869 (2006).
Sejersted, Y. et al. Endonuclease VIII-like 3 (Neil3) DNA glycosylase promotes neurogenesis induced by hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 108, 18802–18807 (2011).
Cortázar, D. et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470, 419–423 (2011).
Kemmerich, K., Dingler, F. A., Rada, C. & Neuberger, M. S. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/− mice. Nucleic Acids Res 40, 6016–6025 (2012).
Chakraborty, A. et al. Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J. Biol. Chem. 290, 24636–24648 (2015).
Rieger, R. A. et al. Characterization of a cross-linked DNA-endonuclease VIII repair complex by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom 11, 505–515 (2000).
Sidorkina, O. M. & Laval, J. Role of the N-terminal proline residue in the catalytic activities of the Escherichia coli Fpg protein. J. Biol. Chem 275, 9924–9929 (2000).
Klungland, A. et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 96, 13300–13305 (1999).
Minowa, O. et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 97, 4156–4161 (2000).
Sakumi, K. et al. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 63, 902–905 (2003).
Xie, Y. et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 64, 3096–3102 (2004).
Sakamoto, K. et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 67, 6599–6604 (2007).
Sampath, H. et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 300, E724–34, doi:10.1152/ajpendo.00387.2010 (2011).
Sampath, H. et al. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS One 7, e51697, doi:10.1371/journal.pone.0051697 (2012).
Mulder, J. E., Turner, P. V. & Massey, T. E. Effect of 8-oxoguanine glycosylase deficiency on aflatoxin B1 tumourigenicity in mice. Mutagenesis 30, 401–409 (2015).
Chan, M. K. et al. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 8, 786–794 (2009).
Karahalil, B., de Souza-Pinto, N. C., Parsons, J. L., Elder, R. H. & Bohr, V. A. Compromised incision of oxidized pyrimidines in liver mitochondria of mice deficient in NTH1 and OGG1 glycosylases. J. Biol. Chem. 278, 33701–33707 (2003).
Ischenko, A. A. & Saparbaev, M. K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 415, 183–187 (2002).
Daviet, S. et al. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair 6, 8–18 (2007).
Shafirovich, V. et al. Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA. J. Biol. Chem. 291, 5309–5319 (2016).
Wang, Z. et al. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 6, e1000951, doi:10.1371/journal.pgen.1000951 (2010).
Vallabhaneni, H. et al. Defective repair of oxidative base lesions by the DNA glycosylase Nth1 associates with multiple telomere defects. PLoS Genet. 9, e1002639, doi:10.1371/journal.pgen1002639 (2013).
Vallabhaneni, H. et al. Defective repair of uracil causes telomere defects in mouse hematopoietic cells. J. Biol. Chem. 290, 5502–5511 (2015).
Hailer, M. K., Slade, P. G., Martin, B. D. & Sugden, K. D. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem. Res. Toxicol. 18, 1378–1383 (2005).
Mangerich, A. et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci.USA 109, E1820–1829 (2012).
Graupner, A. et al. Single cell gel electrophoresis (SCGE) and Pig-a mutation assay in vivo-tools for genotoxicity testing from a regulatory perspective: a study of benzo[a]pyrene in Ogg1(−/−) mice. Mutat. Res. Genet. Toxicol. Environ. Mutagen 772, 34–41 (2014).
Graupner, A. et al. Genotoxic effects of two-generational selenium deficiency in mouse somatic and testicular cells. Mutagenesis 30, 217–225 (2015).
Graupner, A. et al. Gamma radiation at a human relevant low dose rate is genotoxic in mice. Sci. Rep 6, 32977, doi:10.1038/srep32977 (2016).
Phonethepswath, S., Bryce, S. M., Bemis, J. C. & Dertinger, S. D. Erythrocyte-based Pig-a gene mutation assay: demonstration of cross-species potential. Mutat. Res. 657, 122–126 (2008).
Dobrovolsky, V. N., Miura, D., Heflich, R. H. & Dertinger, S. D. The in vivo Pig-a gene mutation assay, a potential tool for regulatory safety assessment. Environ. Mol. Mutagen. 51, 825–835 (2010).
Hernández-Campo, P. M. et al. Quantitative analysis of the expression of glycosylphosphatidylinositol-anchored proteins during the maturation of different hematopoietic cell compartments of normal bone marrow. Cytometry B. Clin. Cytom 72, 34–42 (2007).
McGoldrick, J. P., Yeh, Y. C., Solomon, M., Essigmann, J. M. & Lu, A. L. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell Biol. 15, 989–996 (1995).
Sakumi, K. et al. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 268, 23524–23530 (1993).
Isogawa, A. Functional cooperation of Ogg1 and Mutyh in preventing G: C–>T: a transversions in mice. Fukuoka Igaku Zasshi 95, 17–30 (2004).
Hirano, S. et al. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J. Biol. Chem. 78, 38121–38124 (2003).
Larsen, E. et al. Repair and mutagenesis at oxidized DNA lesions in the developing brain of wild-type and Ogg1−/− mice. Oncogene 25, 2425–2432 (2006).
Bhalli, J. A., Shaddock, J. G., Pearce, M. G. & Dobrovolsky, V. N. Sensitivity of the Pig-a assay for detecting gene mutation in rats exposed acutely to strong clastogens. Mutagenesis 28, 447–455 (2013).
Melis, J. P., van Steeg, H. & Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal 18, 2409–2419 (2013).
Bridge, G., Rashid, S. & Martin, S. A. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers 5, 1597–1614 (2014).
Theriot, C. A., Hegde, M. L., Hazra, T. K. & Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 9, 643–652 (2010).
Hegde, M. L. et al. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 110, E3090–3099 (2013).
Dutta, A., Yang, C., Sengupta, S., Mitra, S. & Hegde, M. L. New paradigms in the repair of oxidative damage in human genome: mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell. Mol. Life Sci. 72, 1679–1698 (2015).
Aamann, M. D. et al. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 135, 1–14 (2014).
Regnell, C. E. et al. Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep 2, 503–510 (2012).
Dertinger, S. D., Bryce, S. M., Phonethepswath, S. & Avlasevich, S. L. When pigs fly: immunomagnetic separation facilitates rapid determination of Pig-a mutant frequency by flow cytometric analysis. Mutat. Res. 721, 163–170 (2011).
O’Callaghan, N. J. et al. A quantitative PCR method for measuring absolute telomere length. Biol. Proced. Online. 13, doi:10.1186/1480-9222-13-3 (2011).
Acknowledgements
This project has been supported by the Research Council of Norway (KS and HN), and through its Centres of Excellence funding scheme, project number 223268/F50 (AKO, AG, JMA); The Norwegian Cancer Society (VR and GAH) and the South Eastern Norway Health Authority (HN, VR). MS analysis was performed by the Proteomics and Metabolomics Core Facility (PROMEC), NTNU, which is founded by the Faculty of Medicine at NTNU and Central Norway Regional Health authority. Costs associated with Prototype MutaFlow Kits were defrayed with a grant from the National Institute of Health/National Institute of Environmental Health Sciences (NIEHS; grant no. R44ES018017). The contents are solely the responsibility of the authors, and do not necessarily represent the official views of the NIEHS.
Author information
Authors and Affiliations
Contributions
V.R., L.L., A.K.O., G.A.H. and M.B. conceived the project and designed the experiments. V.R., L.L., A.K.O., G.A.H., M.B., H.N. and G.S. supervised the project. V.R., L.L., R.S., K.S., C.G.N., Y.E., A.K. (Anna Kuśnierczyk), G.A.H., A.G. and J.M.A. performed all the experimental work. A.K. (Arne Klungland) provided key reagents. V.R. and L.L. drafted the manuscript, which was completed collaboratively by L.L., V.R., G.A.H. and M.B. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rolseth, V., Luna, L., Olsen, A.K. et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci Rep 7, 4384 (2017). https://doi.org/10.1038/s41598-017-04472-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04472-4
This article is cited by
-
RNaseH2A downregulation drives inflammatory gene expression via genomic DNA fragmentation in senescent and cancer cells
Communications Biology (2022)
-
NEIL1 and NEIL2 DNA glycosylases modulate anxiety and learning in a cooperative manner in mice
Communications Biology (2021)
-
Restoration of Cognitive Performance in Mice Carrying a Deficient Allele of 8-Oxoguanine DNA Glycosylase by X-ray Irradiation
Neurotoxicity Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
