
Abstract
All grass leaves are strap-shaped with a series of parallel veins running from base to tip, but the distance between each pair of veins, and the cell-types that develop between them, differs depending on whether the plant performs C3 or C4 photosynthesis. As part of a multinational effort to introduce C4 traits into rice to boost crop yield, candidate regulators of C4 leaf anatomy were previously identified through an analysis of maize leaf transcriptomes. Here we tested the potential of 60 of those candidate genes to alter leaf anatomy in rice. In each case, transgenic rice lines were generated in which the maize gene was constitutively expressed. Lines grouped into three phenotypic classes: (1) indistinguishable from wild-type; (2) aberrant shoot and/or root growth indicating possible perturbations to hormone homeostasis; and (3) altered secondary cell wall formation. One of the genes in class 3 defines a novel monocot-specific family. None of the genes were individually sufficient to induce C4-like vein patterning or cell-type differentiation in rice. A better understanding of gene function in C4 plants is now needed to inform more sophisticated engineering attempts to alter leaf anatomy in C3 plants.
Similar content being viewed by others


Introduction
Genes that regulate developmental processes have traditionally been identified through mutant screens for phenotypic defects. This approach has been enormously powerful for the elucidation of genetic pathways that underpin a large number of developmental mechanisms in model organisms across the phylogenetic range. However, where redundancy or complex genetic interactions operate, such mutant screens have rarely been successful. The development of a characteristic leaf anatomy known as ‘Kranz’ is a case in point. Despite concerted efforts to identify mutations that disrupt Kranz patterning, only two mutants have been identified1, 2 and both of these cause very subtle defects.
Kranz anatomy is a feature of plants that carry out C4 photosynthesis (reviewed in ref. 3). Whereas C3 plants carry out photosynthesis in a single photosynthetic cell-type, most C4 plants compartmentalize photosynthetic reactions between two distinct cell-types known as mesophyll (M) and bundle sheath (BS). For the C4 metabolic pathway to be effective, M cells need to be in contact with BS cells. As such, whereas C3 leaves develop 5–20 M cells between each pair of veins, C4 leaves develop concentric wreaths of BS and M cells around each vein (V) so that each pair of veins is normally separated by four cells in a V-BS-M-M-BS-V unit. This anatomy led to the name Kranz, which is German for wreath4. Remarkably, Kranz anatomy has evolved on over 60 independent occasions5, but there is currently very little indication of the genetic mechanisms that were recruited to enable the transition from the ancestral C3-type leaf anatomy.
Over the last ten years, bioinformatics approaches have increasingly been used to interrogate genome and transcriptome datasets with a view to identifying novel regulators of plant development. In the context of understanding C4 physiology and evolution, many comparisons have been made between different developmental stages within C4 plants6,7,8,9,10,11,12, and between closely related C3 and C4 species13,14,15,16,17,18. However, the most focussed study in terms of understanding how Kranz anatomy develops exploited the fact that the C4 plant maize develops two type of leaves – those with Kranz anatomy (foliar leaves) and those without Kranz anatomy (husk leaves surrounding the female inflorescence)19. The most striking anatomical difference between the two leaf types is that pairs of veins are separated by four cells in foliar leaves and by ~15–20 cells in husk leaves20. Genome-wide comparisons of transcriptomes from developing foliar and husk leaf primordia of maize identified 283 genes as potential positive regulators of Kranz anatomy19, 21.
The power of bioinformatics to generate lists of candidate developmental regulators has been proven. However, gene function can only be validated experimentally. In some model organisms this is straightforward in that loss- and gain- of function manipulations can be carried out both precisely and rapidly (e.g. ref. 22). However, even in the era of genome editing, testing necessity of gene function in the current C4 model systems is not high throughput enough to tackle large gene lists. In the face of this challenge, candidate positive regulators of Kranz anatomy were tested for sufficiency to perturb leaf anatomy in the C3 plant rice. 60 maize genes, most of which encoded transcription factors, were individually transformed into rice under the control of the maize ubiquitin promoter. Phenotypic characterization of vein spacing in leaves of transgenic plants (as a primary indicator of a shift towards Kranz-like anatomy) revealed that none of the candidate genes specifically altered leaf anatomy when expressed constitutively. Indeed, constitutive expression of ~75% of the tested genes had no apparent phenotypic effect on rice growth. The remaining 13 genes conditioned phnetypes that may represent roles in hormone signalling and/or secondary wall formation, providing novel insights into gene function.
Results and Discussion
Constitutive expression of maize genes in rice
To determine whether genes that had been identified in maize as candidate regulators of Kranz anatomy could induce Kranz-like features in the leaves of rice, a systematic transgenic approach was adopted. Of the 283 genes that had previously been identified as putative regulators of Kranz patterning19, 60 genes were chosen for analysis. The main criterion for selection was a predicted regulatory role, and hence the majority of genes encoded transcription factors (36 genes) with another group encoding leucine rich repeat receptor like kinases (LRR-RLK) (14 genes). Sequences for each of the 60 genes were either amplified by reverse transcriptase polymerase chain reaction (RT-PCR), using RNA isolated from maize shoots comprised of the shoot apical meristem plus plastochron (P) 1–5 leaf primordia (a plastochron is the time interval between initiation of primordia at the shoot apex, with P1 primordia being the youngest and closest to the apex), or by PCR from maize genomic DNA. Amplified sequences (Supplementary Information S1) were ligated downstream of the maize ubiquitin promoter in transformation vectors and then constructs were transformed into either indica (IR64) or japonica (Kitaake) rice varieties. Positive transformants were validated for all 60 genes by genomic PCR or DNA blot analysis. Phenotypic analyses were carried out either on multiple independent T0 lines (29 genes) and/or on multiple individuals from 2–3 independent T1 lines (31 genes). The list of genes analyzed is summarized in Table 1.
Constitutive expression of 47 candidate regulators of Kranz anatomy in maize caused no apparent phenotypic defects in rice
To establish a baseline against which perturbations to leaf anatomy could be quantitatively assessed, variation in vein number versus leaf width was first quantified in both IR64 and Kitaake rice varieties. In each case, a regression analysis was carried out using measurements of wild-type leaves at a number of different developmental stages, when grown in different environmental conditions, and in both T0 and T1 null segregants from transformation experiments (Supplementary Dataset S1). In both IR64 and Kitaake varieties, a linear relationship was revealed between leaf width and vein number, a relationship that was conserved regardless of developmental age or environmental growth conditions. Essentially, wider leaves have more veins across the mediolateral leaf axis and narrow leaves have fewer, with the distance between veins remaining roughly equivalent.
T0 lines transformed with 29 of the candidate genes (including 14 encoding transcription factors and 11 encoding receptor kinases) exhibited normal vein spacing patterns, as judged by regression analysis of leaf width versus vein number measurements (Table 1 and Supplementary Dataset S2 and Information S2). T1 lines transformed with a further 18 genes (including 13 encoding transcription factors) similarly exhibited normal vein spacing patterns (Table 1 and Supplementary Dataset S3 and Information S3). Qualitatively, these T0 and T1 lines were phenotypically normal throughout the lifecycle.
ZmIDD16, ZmbHLH106 and ZmHCA2 may influence auxin signalling
Rice transformation protocols rely on the regeneration of plantlets from callus, using an excess of cytokinin to auxin in regeneration media to promote shoot growth. Constitutive expression of three genes prevented plantlets from regenerating under these conditions. Phylogenetic analysis revealed that these genes were: (1) ZmINDETERMINATE DOMAIN (IDD16) (an ortholog of IDD14/15/16 in Arabidopsis23 and LOOSE PLANT ARCHITECTURE1 (LPA1) in rice24) (Supplementary Figure S1); (2) an ortholog of bHLH106 in Arabidopsis25 (Supplementary Figure S2); and (3) an ortholog of Arabidopsis HIGH CAMBIAL ACTIVITY2 (HCA2)26 (Supplementary Figure S3). The maize genes are thus ZmIDD16, ZmbHLH106 and ZmHCA2 (Table 1). Notably, the Arabidopsis orthologs of ZmIDD16 induce the expression of auxin biosynthesis and transport genes23, and although primarily characterized for the ability to confer salt tolerance, AtbHLH106 also activates transcription of the auxin biosynthesis gene AtYUCCA5 25, 27. AtHCA2 induces the formation of interfascicular cambium in Arabidopsis, a process known to be promoted by auxin28, with gain of function mutations leading to ectopic formation of vascular bundles in both stems and leaves26. Together these observations suggest that ZmIDD16, ZmbHLH106 and ZmHCA2 may promote auxin biosynthesis and/or signalling, such that constitutive gene expression in rice callus prevents plantlet regeneration by effectively reducing the cytokinin to auxin ratio. However, other explanations are possible and more direct evidence is needed to confirm the function of each gene.
Constitutive expression of ZmSAUR60, ZmSACL3 or a maize YUCCA gene perturbed shoot and/or root development in rice
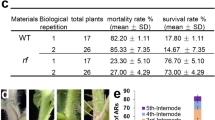
Once transgenic plantlets regenerate from callus, endogenous developmental processes regulate cytokinin to auxin ratios in order to promote shoot and root growth, with cytokinin primarily promoting growth in the shoot meristem and auxin promoting growth in the root. Perturbed shoot and root phenotypes in multiple independent lines constitutively expressing each of three transgenes suggested that gene expression may interfere with cytokinin/auxin homeostasis (Fig. 1). Phylogenetic analysis demonstrated that one of the genes encodes a flavin monoxygenase-like enzyme of the YUCCA family (Supplementary Figure S4). YUCCA genes act in the auxin biosynthesis pathway and the phenotype of rice lines overexpressing the maize YUCCA gene (Fig. 1a,b) is consistent with excessive auxin levels. As previously seen in lines overexpressing rice YUCCA genes29, the maize gene caused a proliferation of curled leaflets and hairy roots (Fig. 1b). None of over 20 T0 plantlets survived transplantation to soil. The second gene had the opposite effect on root growth in that very few, if any, lateral roots were formed (Fig. 1c–h). Phylogenetic analysis revealed that the gene encodes a SMALL AUXIN UPREGULATED RNA (SAUR) protein (Supplementary Figure S5), previously annotated as ZmSAUR60 30. Although not the most closely related rice gene to ZmSAUR60, overexpression of OsSAUR39 has been shown to negatively regulate auxin synthesis and transport in rice31. The phenotype of transgenic lines shown in Fig. 1f–j suggests that ZmSAUR60 may have a similar role, preventing auxin flow into the root and thus inhibiting primary root elongation and lateral root initiation. The third gene that severely perturbed growth in young plantlets was a bHLH transcription factor that is orthologous to SACL3 in Arabidopsis (Supplementary Figure S6). This gene has thus been named ZmSACL3 (Table 1). In Arabidopsis, SACL3 binds to the bHLH protein LONESOME HIGHWAY (LHW), preventing LHW from interacting with another bHLH protein, TARGET OF MONOPTEROS5 (TMO5)32. Given that LHW/TMO dimers activate cytokinin biosynthesis33, 34, competitive inhibition through an excess of SACL3 would lead to reduced levels of cytokinin, and an equivalent phenotype to that seen with increased auxin levels. Although the plantlets overexpressing ZmSACL3 (Fig. 1i–l) reach a more advanced stage than those overexpressing the YUCCA gene (Fig. 1B), in both cases, the severely curled leaves might be explained by a decreased cytokinin to auxin ratio.

Phenotype of transgenic lines that fail to regenerate viable plants. (a,b) T0 plants of control (a) and transgenic (b) lines transformed with a maize YUCCA gene. (c–h) T0 plants of control (c) and transgenic (d–h) lines transformed with ZmSAUR60. (i–l) T0 plants of control (i) and transgenic (j–l) lines transformed with ZmSACL3. Scale bars = 0.5 cm (a,c–h); 0.2 cm (b); 1 cm (i,j,l); 0.8 cm (k).
Perturbations to auxin homeostasis may also underpin the dwarfed phenotype seen in lines expressing an LRR kinase (Table 1 and Fig. 2), but other possibilities cannot be excluded. Transgenic lines grew more slowly than non-transgenic controls but were fertile, exhibited normal vein spacing patterns, and were propagated to the T1 generation. Phylogenetic analysis revealed homology of the transgene to a clade of five genes from Arabidopsis (Supplementary Figure S7), loss of function mutations in which cause resistance to an auxin transport inhibitor (At2G23300 & At5G67280)35 or to abscisic acid (AtRDK1)36.

Transgenic lines that overexpress an LRR kinase are dwarfed. (a) Non-transgenic plants (left hand panel) are taller than transgenic plants (right hand panel). (b) Regression plot showing vein number versus leaf width for one individual from each of two independent T1 lines – 50.1 (orange spot) & 50.2 (blue spot). (c–e) Cropped gel images of genomic (c) and RT- (d,e) PCR reactions illustrate transgene presence and transcript levels in an individual from each T1 line. Scale bar = 5 cm.
A failure to differentiate abaxial sclerenchyma is associated with drooping leaves in rice lines expressing a maize R2R3 MYB gene
Transgenic lines overexpressing a gene encoding an R2R3 MYB transcription factor exhibited drooping, abaxially curled leaves (Table 1 and Fig. 3). Vein spacing and number were unperturbed in the leaves of transgenic plants (Fig. 3c) but transverse leaf sections revealed that intermediate veins often lacked sclerenchyma cell connections to the abaxial epidermis (Fig. 3g). Given the structural role of sclerenchyma, this defect would be sufficient to cause abaxial leaf curling. Phylogenetic analysis revealed that the maize gene is orthologous to LATE MERISTEM IDENTITY 2 (LMI2), a gene that regulates the vegetative to inflorescence transition in Arabidopsis37 (Supplementary Figure S8). No flowering time defects were observed in the transgenic rice lines, however, LMI2 is itself unusual, being nested in a clade that contains many positive and negative regulators of epidermal cell differentiation38. In this context, it is possible that the maize R2R3 MYB functions as a negative regulator of sclerenchyma development.

Transgenic lines overexpressing a maize R2R3 MYB gene exhibit drooping leaves and have fewer abaxial sclerenchyma cells than wild-type. (a) T1 transgenic plants (right hand panel) are shorter than non-transgenic plants (left hand panel), and exhibit a drooping leaf phenotype. (b) T0 plant showing abaxial curling of the flag leaf (white arrow). (c) Regression plot showing vein number versus leaf width for 6 individuals from each of two independent T1 lines − 33.1 (orange spots) & 33.2 (blue spots). (d,e) Cropped gel images of genomic (d) and RT- (e) PCR reactions illustrate transgene presence and transcript levels in representatives of each T1 line. (f,g) Representative transverse leaf sections from wild-type (f) and transgenic line 33.1 (g) plants, showing presence - white arrows in (f), and absence - white arrows in (g), of abaxial sclerenchyma. Scale bar = 5 cm (a); 50 µm (f,g).
Enhanced sclerenchyma formation around leaf veins infers a role for a novel gene in secondary cell wall formation
A novel gene with no functional annotation conditioned a striking phenotype in rice overexpression lines (Table 1 and Figs 4 and S9). Regenerating plants exhibited shorter roots with fewer laterals than non-transgenic siblings (Fig. 4a,b), and shoots were stunted and infertile (Fig. 4c). Vein spacing and number were unperturbed in the leaves of transgenic plants (Fig. 4f), however, transverse leaf sections revealed excessive secondary wall formation around vascular bundles (Fig. 4g–l). Specifically, more sclerenchyma cells (which have lignified cell walls) developed on the adaxial side of major leaf veins and the cells had thicker cell walls than in wild-type plants (Fig. 4g,h,k,l). In addition, one or two BS cells around each vascular bundle often appeared larger than the other cells (Fig. 4h–l).

Transgenic lines overexpressing a protein of unknown function exhibit aberrant lignification around vascular bundles. (a–c) Compared to non-transgenic roots (a) and shoots (c) - left hand panel), growth of transgenic roots (b) and shoots (c)- right hand panel) is stunted. (d,e) Cropped gel images of genomic (d) and RT- (e) PCR reactions illustrate transgene presence and transcript levels in two representative T0 lines. (f) Regression plot showing vein number versus leaf width for 8 independent T0 lines. (g,l) Transverse cross sections of wild-type (g) and transgenic (h–l) leaves showing enhanced number of thicker walled sclerenchyma cells (white arrows) and enlarged and/or ectopic cells around veins (yellow arrows). Coloured circles in (d,e) illustrate corresponding datapoints in (f). Scale bars = 1 cm (a); 8 cm (b); 70 µm (g,h,l); 60 µm (i–k).
Inducible expression of ZmWRKY12 inhibits lobing of mesophyll cells in riceN
Phylogenetic analysis of one of the candidate Kranz regulators revealed orthology to AtWRKY12 in Arabidopsis (Supplementary Figure S10) and thus the maize gene was named ZmWRKY12. Rice callus transformed with ZmWRKY12 driven by the constitutive ubiquitin promoter failed to regenerate, however, callus transformed with an estradiol-inducible transgene regenerated normally in the absence of estradiol and T1 seed were harvested. When T1 seed were germinated in the presence of estradiol, plants were severely dwarfed and had minimal root growth (Fig. 5a). This phenotype was consistent with increased ZmWRKY12 transcript levels (relative to ubiquitin) as compared to untreated control plants (Fig. 5c). Quantitative analysis of vein number versus leaf width in estradiol-treated transgenic plants did not reveal any perturbations, however, transverse leaf sections revealed a mesophyll cell defect (Fig. 5d–i). Mesophyll cells in rice leaves are smaller than those in most other grasses but the cells have extensively lobed walls that effectively increase the surface area for CO2 diffusion39. Nothing is known about how this lobed shape is formed but mesophyll cells in the induced transgenic plants had no lobes (Fig. 5e,g,i). Given that AtWRKY12 is a negative regulator of secondary cell wall formation in Arabidopsis40, it is likely that constitutive expression of ZmWRKY12 prevented callus regeneration through an inability to develop vascular tissue (which requires the formation of secondary walls). More intriguingly, it appears as though the lobing of rice mesophyll cells may require secondary cell wall formation.

Expression of ZmWRKY12 inhibits cell wall growth in rice. (a) Non-transgenic plants (two left hand plants) are taller than plants in which expression of the transgene has been induced by germination on estradiol (two right hand plants). (b,c) Cropped gel images of genomic (b) and RT- (c) PCR reactions illustrate transgene presence in two individuals of T1 line 34.15(i) and enhanced transgene transcript levels (relative to ubiquitin) after exposure (+) of individuals from two independent T1 lines to estradiol. (d–i) Transverse cross sections of non-transgenic (d,f,h) and transgenic (e,g,i) young (d,e) and older (f–i); (h) and (i) are magnified images of (f) and (g) respectively) leaves show smaller, non-lobed mesophyll cells in transgenic plants. Scale bars = 1.5 cm (a); 75 µm (d–g); 25 µm (h,i).
Three maize ZnF homeodomain genes and an endogenous rice ortholog caused spindly growth and lodging when constitutively expressed
Lines that constitutively expressed any of three genes encoding closely related ZnF homeodomain (ZnF-HD) proteins exhibited drooping stems and leaves (Table 1 and Fig. 6a–d,f–k). None of the lines were strong enough to produce T1 seed but quantification of vein number versus leaf width in T0 plants revealed no differences from wild-type (Fig. 6l–n). A spindly shoot phenotype was also observed in an activation tagged line in which a related rice gene (Os03g50920) was ectopically expressed, but in this case T3 populations were obtained. Comparisons between homozygous lines with or without the activation tag revealed a dramatic lodging phenotype at maturity in plants that ectopically expressed the endogenous rice gene (Figs 6e and S11).

Ectopic expression of ZnF homeobox genes promotes spindly growth and lodging. (a–d) Transgenic plants expressing ZmHBc (b), ZmHBa (c) or ZmHBb (d) transgenes are spindly compared to non-transgenic controls (a). (e) Plants ectopically expressing the rice ortholog of ZmHBc exhibit severe lodging at maturity (right). Null segregants of the same age are on the left. (f–k) Cropped gel images of genomic (f,g,h) and RT- (i,j,k) PCR reactions illustrate transgene presence and transgene transcript levels in ZmHBa (f,i), ZmHBb (g,j) and ZmHBc (h,k) lines. (l–n) Regression plots showing vein number versus leaf width for two (l –ZmHBa) and six (m – ZmHBb; n – ZmHBc) individual T0 lines. Coloured circles in (f–k) correspond to datapoints in (l–n). Scale bars = 8 cm (a–d); 12 cm (e).
Phylogenetic analysis using a subset of monocot and eudicot species, and the moss Physcomitrella patens as an outgroup resolved five main clades of flowering plant ZnF-HD genes (Fig. 7) that were consistent with those previously reported for Arabidopsis genes in this family41. Two of the maize genes, referred to here as ZmHBa (GRMZM2G069365) and ZmHBb (GRMZM2G417229), were nested in a clade with AtHB22 and AtHB25 genes. Functional analysis in Arabidopsis revealed that AtHB25 positively regulates GIBBERLLIC ACID3-OXIDASE2 (GA3OX2), leading to increased levels of gibberellic acid (GA) in AtHB25 gain of function lines, and that AtHB25 and AtHB22 act redundantly42. Increased GA levels would explain the spindly phenotype of transgenic rice lines overexpressing ZmHBa and ZmHBb genes (Fig. 6c,d), either through a direct effect of GA on cell expansion or through an inhibition of lignin content (as previously reported in GA2OX overexpression lines of switchgrass)43. The third maize gene, referred to here as ZmHBc (GRMZM2G425236), grouped with AtHB23, AtHB26, AtHB29, AtHB30 and AtHB34. ZmHBc is orthologous to Os03g50920. Although the Arabidopsis orthologs of ZmHBc function in stress responses (where known)44, 45, perturbed GA levels could also explain the spindly phenotype of ZmHBc overexpression lines (Fig. 6b), and the lodging of lines ectopically expressing the corresponding rice ortholog (Fig. 6e).

Phylogenetic tree of Zn finger homeodomain proteins. Maximum-likelihood tree of the ZnF-HD orthogroup (as defined by Orthofinder)56. Numbers at each node are support values based on the consensus tree of 10 bootstrap replicates. Sub-groups are denoted with different line colours. The Arabidopsis proteins (red text) plus the three maize proteins encoded by the genes analysed in this study (green text) are highlighted in yellow.
Summary
Constitutive expression of 60 maize genes in rice has revealed three phenotypic classes. The largest class was phenotypically indistinguishable from wild-type (Table 1 and Supplementary Information S1 and S3). More than 25 genes in this class encoded transcription factors which were expected to alter the expression of a number of downstream genes. The lack of phenotypic perturbation observed suggests either that plant developmental processes are buffered against changes in these particular transcriptional networks or that the maize genes cannot activate downstream targets in rice. Notably one of the genes in this class is ZmSCARECROW1, a gene which when mutated in maize causes subtle defects in Kranz anatomy1. Clearly ZmSCR1 is not sufficient to induce Kranz-like traits in rice, at least not when expressed ubiquitously.
The second phenotypic class altered shoot and/or root growth in ways that inferred perturbed hormone signalling (Figs 1, 2 and 6), although other explanations are possible. In the six cases where gene expression either prevented callus regeneration (Table 1) or led to the regeneration of inviable plantlets (Fig. 1), the endogenous gene expression profile in maize was such that transcript levels were much lower in P5 leaf primordia than in younger P3/4 primordia19. This suggests that repression of gene expression at P5 is necessary for normal maize leaf development. Constitutive expression in transgenic rice plants might thus be expected to perturb shoot development. Notably a number of genes in this class are orthologs of genes involved in auxin biosynthesis or signalling in Arabidopsis, and the phenotype of transgenic lines is consistent with a similar role for the maize genes. Loss of function mutants need to be identified to confirm or refute this suggestion.
Phenotypes in the final class were associated with perturbed secondary wall formation (Figs 3, 4 and 5). Given that secondary wall formation is crucial during early leaf development it is not surprising that such genes were identified as potential regulators of C4 patterning. However, the gain of function phenotypes resulting from constitutive expression raise interesting questions about potential roles in regulating cellular differentiation more broadly. For example, the gain of function phenotype for one of these genes, ZmWRKY12, revealed a role for secondary cell walls in the formation of lobed mesophyll cells in rice (Fig. 3). This trait is characteristic of rice mesophyll cells but until now nothing was known about how lobing was achieved. Just as ZmWRKY2 inhibited mesophyll cell lobing, constitutive expression of an R2R3 Myb gene suppressed sclerenchyma formation around veins (Fig. 4). A third gene defined a novel gene family which is monocot-specific. Gain of function in this case led to the formation of ectopic cell-types around veins (Fig. 5). These cells resembled extra sclerenchyma, but could possibly be bulliform-type cells. Unfortunately, there are no markers to distinguish these two cell-types. Further functional insight into the role of all three genes in cell-type differentiation requires the generation of loss of function mutants.
The development of Kranz anatomy in C4 plants is complex, minimally requiring altered leaf venation and cell-type differentiation in comparison to leaf development in ancestral C3 plants. Despite this complexity, the trait has evolved on over 60 independent occasions5. In this study we tested whether potential regulators of Kranz anatomy in maize were individually sufficient to alter leaf anatomy in rice when constitutively expressed. Although none of the genes elicited a shift towards Kranz-type venation patterning, in some cases cellular differentiation was perturbed. In other cases, phenotypic perturbations were so severe that potentially more subtle effects were obscured by the coarse nature of constitutive expression experiments. In these cases, further analyses of gene function need to be carried out (both necessity and sufficiency), exploiting different promoters for transgenic experiments (both inducible and cell-type specific), and also analyzing combinations of gene function. Importantly, as efforts continue to try and engineer rice leaf anatomy, 47 genes can be eliminated from further study because even with constitutive expression, no impact on rice leaf development was observed.
Methods
Rice germplasm
Transgenic lines were generated using Oryza sativa indica cultivar IR64 or japonica cultivar Kitaake. Activation tagged lines were previously generated in Tainung 67 and are part of the Taiwan Rice Insertional Mutant (TRIM) collection http://trim.sinica.edu.tw/ 46.
Plant growth conditions
Kitaake and Tainung 67 lines were grown in soil (John Innes Compost No.2) in a transgenic greenhouse in Oxford. Day/night temperature was maintained at 30 °C/22 °C ± 3 °C with a diurnal light regime of 16 h light (supplemented to ~300 μM m−2 sec−1) and 8 h dark. IR64 lines were grown in soil in 7 L pots in a transgenic screenhouse under natural light conditions (maximum light intensity up to 2000 μM m−2 sec−1 on a sunny day) at the International Rice Research Institute, Los Banos, Philippines where day/night temperatures were 35 °C/28 °C ± 3 °C.
DNA and RNA extraction
Genomic DNA from maize B73 or Kitaake rice lines was isolated using a modified CTAB method47. Genomic DNA from IR64 rice lines was isolated using a potassium acetate method48. Rice leaf RNA was isolated using TRIZOL reagent (Invitrogen) and maize leaf RNA using either TRIZOL or a mirVana™ miRNA isolation kit (Applied Biosystems), as described in ref. 19.
Generation of transformation constructs
Coding sequences corresponding to each of the candidate genes was isolated by PCR amplification using Phusion High-Fidelity DNA Polymerase (Thermo Scientific) and Gateway® compatible primers (Supplementary Dataset S4). The template for all reactions was maize cDNA that had been generated using RNA isolated from P1-5 leaf primordia and a Transcriptor High Fidelity cDNA Synthesis Kit (Roche). For some genes, the same PCR conditions were used to amplify sequences from genomic DNA. The amplified sequences were subcloned into the Gateway® donor vectors pDONR™ 207 or pENTR/D-TOPO in a BP reaction. The resultant entry clones were sequenced, and the target sequences were then cloned in a LR reaction downstream of the maize ubiquitin promoter (ZmUbipro), into a destination vector modified from pVec8-GFP49 or into the destination vector pSC310. pSC310 vector was created by Sarah Covshoff and kindly gifted to us by Julian Hibberd (University of Cambridge, Cambridge, UK). Estradiol inducible constructs were generated by an LR reaction between entry clones and the destination vector pMDC750. All constructs were given a construct ID and a ‘JL’ gene ID.
Rice transformation
Callus induced from mature rice seeds was used for transformation of the Kitaake cultivar with Agrobacterium tumefaciens strain EHA105 carrying the construct of interest. Callus induction, transformant selection and seedling regeneration were performed at 32 °C under continuous light according to a protocol modified from51 (available to download from https://langdalelab.files.wordpress.com/2015/07/kitaake_transformation_2015.pdf). Hygromycin resistant T0 seedlings that confirmed positive for transgene presence by PCR screening (see below) were transplanted into soil in 0.73 L pots. For IR64, immature embryos were used for agrobacterium mediated transformation using Agrobacterium tumefaciens strain LBA4404 carrying the construct of interest. Callus induction, selection of transformed callus and plantlet regeneration were performed at 30 °C under continuous light according to a protocol modified from52.
Estradiol induced gene expression
For transgene expression analysis, 4th leaves from plants of the inducible lines JL34.15 and JL34.25 were detached and allowed to take up 2 μM β-estradiol or DMSO mock solution by transpiration. Total RNA was extracted after 24 h of treatment at 28 °C in the light, and RT-PCR carried out (see below). For phenotypic characterization of estradiol inducible lines, seeds were germinated alongside wild-type controls on filter paper wetted with 2 μM β-estradiol solution. After germination, plants were cultured in 1/2 MS liquid medium with the same β-estradiol concentration, and images were taken 10 days after germination. Liquid cultures were grown in a growth cabinet with cycles of 28 °C/16 h light and 23 °C/8 h dark.
Genomic PCR screening and RT-PCR
For Kitaake lines, regenerated T0 plants and T1 seedlings (2–3 weeks after germination) were subjected to genomic PCR using primers specific to the cloning vector: pVec8F (TTTAGCCCTGCCTTCATACG, located in the ZmUBI pro ), and pVec8R (ATTGCCAAATGTTTGAACGA, located in the nos terminator). PCR amplification was performed in a total reaction volume of 10 µl containing 5 ul 2xGoTaq® mix (Promega) and 2.5 ul 4 M betaine. PCR conditions were: 95 °C for 5 min; 28 cycles of 95 °C for 30 s, 55 °C for 40 s, 72 °C for 2.5 min; and 72 °C for 5 min. For IR64 lines, regenerated T0 plants and T1 seedlings (2 weeks after germination) were subjected to genomic PCR using gene specific primers (Supplementary Dataset S4). PCR amplification was performed in a total reaction volume of 10 µl containing 5 µl 2xKAPA Plant PCR buffer, 0.1 µl KAPA3G Plant DNA polymerase (KAPABIOSYSTEMS Inc.) and 3.1 µl distilled water. PCR conditions were: 95 °C for 5 min; 32 cycles of 95 °C for 20 s, 60 °C for 15 s, 72 °C for 1 min; and 72 °C for 1 min.
Unless otherwise noted, gene expression analysis was carried out using RNA extracted from fully expanded 4th leaf tissue. Total RNA was treated with RQ1 RNAse free DNAse (Promega, USA) and cDNA was synthesized using Superscript® III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Primers specific to the gene of interest (Supplementary Dataset S4) and 2xGoTaq® mix (Promega) were used in a 10 µl PCR reaction volume to detect the expression of transgenes and/or endogenous rice genes. All PCR products were detected by agarose gel electrophoresis using standard protocols53.
DNA gel blot analysis
For each sample, 6–8 µg of genomic DNA was digested with a restriction endonuclease that had a single cut site in the construct (37 °C for 12–16 h). Digested DNA samples were electrophoresed overnight at 25 volts on a 0.8% agarose gel in 1X TAE buffer and then blotted onto Hybond N+ membrane (GE Healthcare, UK) overnight using 20x SSC as transfer buffer. Blots were hybridised with a digoxigenin (DIG) labeled fragment of the ZmUbi promoter that was synthesised using the PCR DIG Probe Synthesis Kit (Roche Diagnostics, Germany). DNA hybridisation signals were detected using CDP-Star (Roche Diagnostics, Germany).
Quantitative RT-PCR
PCR amplification was carried out using GoTaq Hot Start polymerase (Promega) and amplification detected with 1/60,000 SYBR Green II (Sigma-Aldrich) and the Mx3000 P QPCR System (Agilent). The thermal profile ran as follows: 95 °C, 10 minutes; (95 °C, 15 seconds, 60 °C, 30 seconds, 72 °C, 30 seconds) × 45 cycles. Three technical replicates were carried out per sample and transcript abundance was normalized to the endogenous rice UBIQUITIN gene (Os03g13170).
Phenotypic analysis
Phenotypic analysis was carried out on the expanded 6th leaf of the first tiller. Leaf width and length plus stomatal number on adaxial and abaxial leaf surfaces were first measured and then fresh segments were hand cut from the middle of the leaf blade. For Kitaake lines, segments were embedded in 5%(w/v) agar and sectioned at 70–80 μm with a Vibratome Series 1000 Sectioning System. Sections were viewed and photographed with a Leica DMRB microscope. For IR64 lines, segments were fixed in 2.5 glutaraldehyde, cleared and then stained with Toluidine blue. Hand sections were captured using an Olympus BX51 microscope with a DP71 camera. The cell number between a pair of intermediate veins positioned between the second and third lateral vein in from the leaf margin was recorded for each sample, and the presence or absence of large BS chloroplasts and of M cell invaginations was also noted.
Quantification of vein spacing
The number of veins across the medio-lateral leaf axis was counted either using the Leica DMRB microscope and transverse cross-sections, or using a stereomicroscope and photomicrographs of the leaf surface. 2–5 leaf samples were quantified for each line. Least-squares regression lines of leaf width versus vein number were plotted for both the Kitaake and IR64 datasets using the R stats package54. To maximize the R-squared value, a second order polynomial line was fitted to the Kitaake data, whilst a linear model was used for IR64 data. Data and regression lines were visualized using the R ggplot2 package55.
Phylogenetic analysis
OrthoFinder56 was run over proteome-wide protein sequences of eleven species to identify the groups of orthologous genes (OrthoGroups (OGs)) across them. Species included the eudicots Arabidopsis thaliana and Solanum lycopersicum, the monocots Zea Mays, Sorghum bicolor, Setaria italica, Setaria viridis, Dichanthelium oligosanthes, Oryza sativa, Brachypodium distachyon, the basal angiosperm Amborella trichopoda, and the moss Physcomitrella patens. The OGs which contained the maize genes of interest were selected, and then protein sequences were aligned by MAFFT-lins57. For the ZnF HD phylogeny (Fig. 7), three sequences were removed from the dataset before alignment (D. oligosanthes OEL17202.1 & OEL35852.1, plus A. trichopoda scaffold00011.170) because the genome annotation appeared spurious. Maximum Likelihood phylogenetic trees for each OG were produced from these alignments using IQ-TREE software58, and then consensus trees were finally generated by SUMTREES59 with the Maximum Clade Credibility Topology (MCCT) algorithm.
Accession codes
All accession codes are included in Table 1, and complete sequences are included in Supplementary Information S1.
References
Slewinski, T. L., Anderson, A. A., Zhang, C. & Turgeon, R. Scarecrow plays a role in establishing kranz anatomy in maize leaves. Plant Cell Physiol. 53, 2030–2037 (2012).
Slewinski, T. L. et al. Short-root1 plays a role in the development of vascular tissue and Kranz anatomy in maize leaves. Mol. Plant 7, 1388–1392 (2014).
Langdale, J. A. C4 cycles: past, present, and future research on C4 photosynthesis. Plant Cell 23, 3879–3892 (2011).
Haberlandt, G. Physiologische Pflanzenanatomie. (Wilhelm Engelman, 1896).
Sage, R. F., Christin, P.-A. & Edwards, E. J. The C4 plant lineages of planet Earth. J. Exp. Bot. 62, 3155–3169 (2011).
Wang, L. et al. Comparative analyses of C4 and C3 photosynthesis in developing leaves of maize and rice. Nat. Biotechnol. 32, 1158–65 (2014).
Li, P. et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 42, 1060–1067 (2010).
Pick, T. R. et al. Systems analysis of a maize leaf developmental gradient redefines the current C4 model and provides candidates for regulation. Plant Cell 23, 4208–4220 (2011).
Liu, W.-Y. et al. Anatomical and transcriptional dynamics of maize embryonic leaves during seed germination. Proc. Natl. Acad. Sci. 110, 3979–3984 (2013).
Li, Y. et al. Developmental genetic mechanisms of C4 syndrome based on transcriptome analysis of C3 cotyledons and C4 assimilating shoots in Haloxylon ammodendron. PLoS One 10 (2015).
Yu, C. P. et al. Transcriptome dynamics of developing maize leaves and genomewide prediction of cis elements and their cognate transcription factors. Proc Natl Acad Sci USA 112, E2477–86 (2015).
Lauterbach, M. et al. C3 cotyledons are followed by C4 leaves: intra-individual transcriptome analysis of Salsola soda (Chenopodiaceae). J. Exp. Bot. 68, 161–176 (2017).
Brautigam, A. et al. An mRNA blueprint for C4 photosynthesis derived from comparative transcriptomics of closely related C3 and C4 species. Plant Physiol. 155, 142–156 (2011).
Gowik, U., Brautigam, A., Weber, K. L., Weber, A. P. & Westhoff, P. Evolution of c4 photosynthesis in the genus flaveria: how many and which genes does it take to make c4? Plant Cell 23, 2087–2105 (2011).
Aubry, S., Kelly, S., Kumpers, B. M., Smith-Unna, R. D. & Hibberd, J. M. Deep evolutionary comparison of gene expression identifies parallel recruitment of trans-factors in two independent origins of C4 photosynthesis. PLoS Genet. 10, e1004365 (2014).
Rao, X. et al. Comparative cell-specific transcriptomics reveals differentiation of C4 photosynthesis pathways in switchgrass and other C4 lineages. J. Exp. Bot. 67, 1649–1662 (2016).
Ding, Z. et al. Identification of photosynthesis-associated C4 candidate genes through comparative leaf gradient transcriptome in multiple lineages of C3 and C4 species. PLoS One 10 (2015).
Kümpers, B. M. C. et al. Shared characteristics underpinning C4 leaf maturation derived from analysis of multiple C3 and C4 species of Flaveria. J. Exp. Bot. 68, 177–189 (2017).
Wang, P., Kelly, S., Fouracre, J. P. & Langdale, J. A. Genome-wide transcript analysis of early maize leaf development reveals gene cohorts associated with the differentiation of C4 Kranz anatomy. Plant J. 75, 656–670 (2013).
Langdale, J. A., Rothermel, B. A. & Nelson, T. Cellular patterns of photosynthetic gene expression in developing maize leaves. Genes Dev. 2, 106–115 (1988).
Fouracre, J. P., Ando, S. & Langdale, J. A. Cracking the Kranz enigma with systems biology. J. Exp. Bot. 65, 3327–3339 (2014).
Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science (80-.). 353 (2016).
Cui, D. et al. The Arabidopsis IDD14, IDD15, and IDD16 Cooperatively Regulate Lateral Organ Morphogenesis and Gravitropism by Promoting Auxin Biosynthesis and Transport. PLoS Genet. 9 (2013).
Wu, X., Tang, D., Li, M., Wang, K. & Cheng, Z. Loose Plant Architecture1, an INDETERMINATE DOMAIN protein involved in shoot gravitropism, regulates plant architecture in rice. Plant Physiol. 161, 317–29 (2013).
Ahmad, A. et al. BHLH106 integrates functions of multiple genes through their g-box to confer salt tolerance on Arabidopsis. PLoS One 10 (2015).
Guo, Y., Qin, G., Gu, H. & Qu, L.-J. Dof5.6/HCA2, a Dof transcription factor gene, regulates interfascicular cambium formation and vascular tissue development in Arabidopsis. Plant Cell 21, 3518–3534 (2009).
Woodward, C. et al. Interaction of auxin and ERECTA in elaborating Arabidopsis inflorescence architecture revealed by the activation tagging of a new member of the YUCCA family putative flavin monooxygenases. Plant Physiol. 139, 192–203 (2005).
Suer, S., Agusti, J., Sanchez, P., Schwarz, M. & Greb, T. WOX4 imparts auxin responsiveness to cambium cells in Arabidopsis. Plant Cell 23, 3247–59 (2011).
Yamamoto, Y., Kamiya, N., Morinaka, Y., Matsuoka, M. & Sazuka, T. Auxin biosynthesis by the YUCCA genes in rice. Plant Physiol. 143, 1362–1371 (2007).
Chen, Y., Hao, X. & Cao, J. Small auxin upregulated RNA (SAUR) gene family in maize: Identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J. Integr. Plant Biol. 56, 133–150 (2014).
Kant, S., Bi, Y. M., Zhu, T. & Rothstein, S. J. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 151, 691–701 (2009).
Vera-Sirera, F. et al. A bHLH-Based Feedback Loop Restricts Vascular Cell Proliferation in Plants. Dev. Cell 35, 432–443 (2015).
De Rybel, B. et al. Plant development. Integration of growth and patterning during vascular tissue formation in Arabidopsis. Science 345, 1255215 (2014).
Ohashi-Ito, K. et al. A bHLH complex activates vascular cell division via cytokinin action in root apical meristem. Curr. Biol. 24, 2053–2058 (2014).
ten Hove, C. A. et al. Probing the roles of LRR RLK genes in Arabidopsis thaliana roots using a custom T-DNA insertion set. Plant Mol. Biol. 76, 69–83 (2011).
Kumar, D. et al. ARABIDOPSIS THALIANA RECEPTOR DEAD KINASE1 Functions as a Positive Regulator in Plant Responses to ABA. Mol. Plant doi:10.1016/j.molp.2016.11.011.
Pastore, J. J. et al. LATE MERISTEM IDENTITY2 acts together with LEAFY to activate APETALA1. Development 138, 3189–3198 (2011).
Brockington, S. F. et al. Evolutionary analysis of the MIXTA gene family highlights potential targets for the study of cellular differentiation. Mol. Biol. Evol. 30, 526–540 (2013).
Sage, T. L. & Sage, R. F. The Functional Anatomy of Rice Leaves: Implications for Refixation of Photorespiratory CO2 and Efforts to Engineer C4 Photosynthesis into Rice. Plant Cell Physiol. 50, 756–772 (2009).
Wang, H. et al. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc. Natl. Acad. Sci. USA 107, 22338–43 (2010).
Tan, Q. K.-G. & Irish, V. F. The Arabidopsis zinc finger-homeodomain genes encode proteins with unique biochemical properties that are coordinately expressed during floral development. Plant Physiol. 140, 1095–1108 (2006).
Bueso, E. et al. ARABIDOPSIS THALIANA HOMEOBOX 25 uncovers a role for gibberellins in seed longevity. Plant Physiol. 164, 999–1010 (2013).
Wuddineh, W. A. et al. Identification and overexpression of gibberellin 2-oxidase (GA2ox) in switchgrass (Panicum virgatum L.) for improved plant architecture and reduced biomass recalcitrance. Plant Biotechnol. J. 13, 636–647 (2015).
Choi, H. et al. The homeodomain-leucine zipper ATHB23, a phytochrome B-interacting protein, is important for phytochrome B-mediated red light signaling. Physiol. Plant. 150, 308–320 (2014).
Tran, L. S. P. et al. Co-expression of the stress-inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J. 49, 46–63 (2007).
Lo, S.-F. et al. Genetic resources offer efficient tools for rice functional genomics research. Plant. Cell Environ. 39, 998–1013 (2016).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8, 4321–5 (1980).
Guillemaut, P. & Maréchal-Drourd, L. Isolation of plant DNA: a fast, inespensive, and reliable method. Plant Mol. Biol. Report. 10, 60–65 (1992).
Kim, C. M. & Dolan, L. ROOT HAIR DEFECTIVE SIX-LIKE Class I Genes Promote Root Hair Development in the Grass Brachypodium distachyon. PLoS Genet. 12 (2016).
Curtis, M. D. & Grossniklaus, U. A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133, 462–469 (2003).
Toki, S. et al. Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J. 47, 969–976 (2006).
Hiei, Y. & Komari, T. Improved protocols for transformation of indica rice mediated by Agrobacterium tumefaciens. Plant Cell. Tissue Organ Cult. 85, 271–283 (2006).
Sambrook, J., Fritsch, E. F., Maniatis, T. & Rich, E. F. Molecular cloning: a laboratory manual. (Cold Spring Harbor Press, 1982).
Team, R. R Development Core Team. R A Lang. Environ. Stat. Comput. 55, 275–286 (2015).
Wickham, H. ggplot2. Elegant Graphics for Data Analysis doi:10.1007/978-0-387-98141-3 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274 (2015).
Sukumaran, J. & Holder, M. T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Acknowledgements
We are grateful to the following for technical support: Sayuri Ando, Julie Bull, Zara Lewis (Oxford); Arnelyn Doloiras, Albert de Luna, Leanilyn Lim, Abigail Mabilangan, Maricar Mercado, Florencia Montecillo, Juvy Reyes, Menard dela Rosa (IRRI), and to John Baker for photography. Research in all three institutions was funded by a C4 Rice Project grant from the Bill & Melinda Gates Foundation to IRRI (2012–2015) and to the University of Oxford (2015–2019); M.L.S. & J.A.L. were also funded by the EU FP7 3to4 project. T.H. and O.S. were supported by graduate scholarships from the Newton Abraham (Oxford) and Clarendon (Oxford) Funds respectively. S.Ke. is supported by a Royal Society University Research Fellowship.
Author information
Authors and Affiliations
Contributions
Experimental concept and design – P.W., J.F., J.A.L.; generation of transformation constructs, genotypic and phenotypic analysis of Kitaake lines – P.W., M.L.S., T.H., J.P.F., O.S.; generation of IR64 lines, genotypic and phenotypic analysis of IR64 lines – S.K., A.K.B., H.-C.L., M.J.D., G.R., X.Y., A.B., W.P.Q.; analysis of rice TRIM line – S.-F.L., S.-M.Y.; phylogenetic analysis – B.A.J., S.Ke.; manuscript preparation – J.A.L.; data analysis and interpretation, editing of final manuscript – all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, P., Karki, S., Biswal, A.K. et al. Candidate regulators of Early Leaf Development in Maize Perturb Hormone Signalling and Secondary Cell Wall Formation When Constitutively Expressed in Rice. Sci Rep 7, 4535 (2017). https://doi.org/10.1038/s41598-017-04361-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04361-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
