
Inspired by nature, herein we designed a novel construction of SnO2 anodes with an extremely high lithium storage performance. By utilizing small sheets of graphene oxide, the partitioned-pomegranate-like structure was constructed (SnO2@C@half-rGO), in which the porous clusters of SnO2 nanoparticles are partially supported by reduced graphene oxide sheets while the rest part is exposed (half-supported), like partitioned pomegranates. When served as anode for lithium-ion batteries, SnO2@C@half-rGO exhibited considerably high specific capacity (1034.5 mAh g−1 after 200 cycles at 100 mA g−1), superior rate performance and remarkable durability (370.3 mAh g−1 after 10000 cycles at 5 A g−1). When coupled with graphitized porous carbon cathode for lithium-ion hybrid capacitors, the fabricated devices delivered a high energy density of 257 Wh kg−1 at ∼200 W kg−1 and maintained 79 Wh kg−1 at a super-high power density of ∼20 kW kg−1 within a wide voltage window up to 4 V. This facile and scalable approach demonstrates a new architecture for graphene-based composite for practical use in energy storage with high performance.
Similar content being viewed by others


Introduction
With the rapid evolution of new automobile propulsion and the continuous lightening of electronic devices, the development of lithium-ion batteries (LIBs) toward high energy density, high power density, and long cycling life has been put under urgent. As one of the most important issues in LIBs, new generations of anode materials have been explored, aimed to transcend the theoretical limit of conventional graphite anodes (372 mAh g−1). Besides the novel nanostructured carbon materials such as multimodal porous carbon1, 2, hollow core carbon spheres3, and mesoporous carbon nanofibers4, two kinds of substances are of great interest for their high theoretical capacities: one kind being reversibly lithiated/delithiated through conversion reaction (Fe2O3, Fe3O4, MnO2, CoO, etc.), and the other through alloying reaction (Si, Ge, Sn, SnO2, etc.)5,6,7,8,9,10,11,12. Unfortunately, the pulverization and loss of electrical contact caused by the significant volume change during lithiation/delithiation reaction have been identified as main reasons for the capacity fading of these new anodes10, 11, 13, 14.
Graphene, mostly reduced graphene oxide (rGO), has been widely used to overcome the flaw of various new-type anode materials6,7,8,9, 15,16,17. The ultimate electrical conductivity, ultrahigh specific surface area, and unique two-dimensional shape of graphene exert substantial effects on both the physicochemical property and the nanostructure of these anodes. Packaging nanoparticles of anode materials into clusters and encapsulating the clusters with rGO sheets has been demonstrated as an effective approach for these materials such as Fe3O4, ZnMn2O4 and SnO2 6, 12, 18. Wang et al. prepared ZnMn2O4 porous spheres and encapsulated them with GO18. The composite anode exhibited a capacity of 926.4 mAh g−1 at current density of 200 mA g−1. It is believed that the electrical conductivity of the composite electrode can be improved by rGO, and the voids between the nanoparticles can buffer volume change and prevent agglomeration and pulverization. Unfortunately, when the ZnMn2O4/rGO anode was tested at 1 A g−1, a decline tendency was found only after 120 cycles. The reason may lie in the fact that the encapsulated rGO shell would hinder the rapid penetration and transfer of lithium ions (Li+)19, 20. It is still a great challenge to design novel structures of graphene-based composite anodes to afford high transfer speed of Li+ as well as high reversible capacity and stability, which also accords with the purpose of applying these new-type anode materials in lithium-ion hybrid capacitors (LIHCs) to get energy storage devices with high power density21.
In fact, nature provides us inspiration for the design of new materials. Learning from nature, Cui et al. designed a pomegranate-like anode, where single-phased silicon nanoparticles are encapsulated by a conductive carbon layer that leaves enough room for expansion and contraction during charge-discharge process. The anode showed an excellent cycle life (97% capacity retention after 1000 cycles), high coulombic efficiency (99.87%) and volumetric capacity (1270 mAh cm−3)22. The naturally formed structure of pomegranates is an impressed model in the design of anode materials to accommodate a large volume expansion. Taking advantage of the unique properties of graphene together with the fantastic pomegranate structure, we recently synthesized an anode material based on SnO2, where mesoporous SnO2 clusters were encapsulated by amorphous carbon layers and then wrapped with rGO sheets. The obtained materials exhibited a high reversible capacity of 924 mAh g−1 at 100 mA g−1. However, the encapsulated dual thick carbon shell hindered the rapid penetration and transfer of Li+ and resulted in an unsatisfactory rate performance (240 mAh g−1 at 3 A g−1)12.
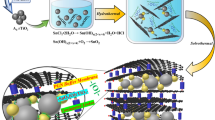
Imitating the structure of partitioned pomegranates, herein we pioneered a brand new structure for extremely high-performance SnO2-rGO composite anode, which can be applied in both LIBs and LIHCs (Fig. 1a). Small sheets of graphene oxide (SGO) were used to construct this delicate structure of SnO2 anodes (SnO2@C@half-rGO), in which the porous clusters of SnO2 nanoparticles are partially supported by rGO sheets and partially exposed. The SnO2 nanoparticles inside the porous clusters are individually covered by thin amorphous carbon shells. The structure in whole is just like partitioned pomegranates. Such a design has multiple advantages: (1) the thin amorphous carbon shell not only limits most SEI formation to the outer surface, but also enables the fast diffusion of Li+; the void space inside the clusters is well defined and evenly distributed around each nanoparticle, which can buffer the expansion of the nanoparticles; (2) rGO provides a conducting framework. Compared with the wholly-wrapped control sample (SnO2@C/rGO, Fig. 1a) prepared using large sheets of graphene oxide (LGO), SnO2@C@half-rGO can maintain the conductive matrix of rGO while open the rapid access for Li+.

(a)Graphical synthesis route of SnO2@C@half-rGO and SnO2@C/rGO. (b,c) TEM images of the SnO2 nanoparticles. (d,e) SEM images of SnO2@C@half-rGO.
As the result, SnO2@C@half-rGO gained a much improved performance as the anode for LIBs. It showed a high reversible capacity up to 1034.5 mAh g−1 at 100 mA g−1 and superior rate performance and stability (370.3 mAh g−1 after 10000 cycles at 5 A g−1). When coupled with graphitized porous carbon (gpC) cathode into LIHCs, the obtained SnO2@C@half-rGO//gpC device exhibited a considerably high energy density of 257 Wh kg−1 at ∼200 W kg−1, and 79 Wh kg−1 was maintained at a super-high power density of ∼20 kW kg−1.
Results
The synthesis route of SnO2@C@half-rGO is schematically represented in Fig. 1a. Firstly, the SnO2 nanoparticles were prepared as hydrosol. TEM images (Fig. 1b,c) of the SnO2 nanoparticles in hydrosol show a well-dispersed morphology, with a uniform size of ∼3 nm. The block copolymer surfactant P123 was mixed with the SnO2 hydrosol to gather the nanoparticles with ample amphiphilic groups, followed by the addition of glucose, which acted as both a reductant23 and the precursor of amorphous carbon. Subsequent introduction of small sheet GO (denoted as SGO) into the system followed by a hydrothermal process led to the reduction of SGO and the figuration of the target structure (SnO2@C@half-rGO).
The wrinkled and porous network of rGO in SnO2@C@half-rGO is manifested in the SEM image (Fig. 1d), while the nested secondary structure is shown in the magnified one (Fig. 1e). It can be clearly identified that the well-formed SnO2 clusters are supported by rGO sheets on part of the cluster surface (half-supported, for short). The element mapping images of Sn, C, and O (Supplementary Fig. S1) indicate a uniform distribution of SnO2 clusters on the rGO sheets in large scale.
The detailed cluster structure of the SnO2 nanoparticles in TEM is clearly shown in Fig. 2a, while the void space spreads among the SnO2 particles. The diffraction rings rather than spots are observed on the SAED pattern (Fig. 2b), which is consistent with the ultra-small size of the SnO2 determined by XRD pattern (Fig. 3a). Moreover, two rings of the (100) and (002) lattice planes of rGO can be found, well corresponding to the flexuose stripe fingerprint of graphitic carbon in the HRTEM image (Fig. 2c). The rGO sheets are found to adhere tightly to the SnO2 clusters. In the magnified area of the SnO2 clusters (Fig. 2d), the lattice fringes of SnO2 can be captured more clearly, while amorphous substance is observed all around the SnO2 nanoparticles, which is inferred to be the amorphous carbon derived from glucose.

TEM image of SnO2@C@half-rGO. (a) The rGO-half-supported clusters in SnO2@C@half-rGO. (b) The SAED pattern of SnO2@C@half-rGO. (c) Magnified image on the edge of the clusters, showing the flexuose stripe of rGO lattice. (d) HRTEM of the SnO2 nanoparticles in SnO2@C@half-rGO.

(a) XRD patterns of LGO, SGO, SnO2 nanoparticles, SnO2@C@half-rGO, SnO2@C/rGO, and SnO2/C. The standard peak position and intensity of PDF#41-1445 SnO2 are shown as verticle lines. (b) Raman spectra of LGO, SGO, SnO2@C@half-rGO, and SnO2@C/rGO. (c) Nitrogen adsorption/desorption isotherms and (d) pore size distribution of SnO2@C@half-rGO, SnO2@C/rGO and SnO2/C.
GO has long been applied as structure framework and conductivity reinforcement in various composites with nanoarchitectonic design for various application24. Generally, GO with micron-scale size was adopted. In this research, the use of SGO (with average lateral size of 0.42 μm) is a key point to construct this partitioned-pomegranate-like composite. The difference in the size and chemical properties of SGO and large sheet GO (named as LGO, the detailed information supplied in Supplementary Figs S2 and S3) can result in different nanostructures when they are introduced to fabricate composites with nanoparticles. While SGO can lead to a half-supported structure in SnO2@C@half-rGO, LGO can and will wholly wrap the SnO2 clusters, which was clearly observed in SEM and TEM (Supplementary Fig. S4).
The wholly-wrapped SnO2 clusters was abbreviated as SnO2@C/rGO and studied as a comparison sample (shown in Fig. 1a). In addition, another control sample was prepared with only SnO2 nanoparticles and amorphous carbon without GO, which was named SnO2/C (Supplementary Fig. S5).
The cassiterite phase of SnO2 was well maintained in SnO2@C@half-rGO, SnO2@C/rGO and SnO2/C after the synthesis, as confirmed by XRD patterns (Fig. 3a). The SnO2 grains in SnO2/C possess the largest size (6.00 nm, Supplementary Table S1), while the ones in SnO2@C@half-rGO and SnO2@C/rGO are smaller (4.91 and 4.63 nm, respectively). It is concluded that both SGO and LGO have inhibition effect on the grain growth of the SnO2 nanoparticles during the hydrothermal process or calcination. The reduction of GO in SnO2@C@half-rGO and SnO2@C/rGO was confirmed by Raman spectroscopy (Fig. 3b). Typical D band (∼1350 cm−1) and G band (∼1590 cm−1) were observed for all the GO-contained samples. As G band is the signal of sp2 carbon while D band is usually regarded as an indication of the disorder in GO or the existence of amorphous carbon, the intensity ratio of D and G (denoted as I D /I G ) is usually calculated to indicate the extent of reduction of GO25, 26. The I D /I G values of SnO2@C@half-rGO and SnO2@C/rGO were determined to be 0.89 and 0.82, respectively, which are both lower than those of SGO (0.98) and LGO (0.91), despite the influence of the amorphous carbon in both samples. This decrease of I D /I G demonstrates the successful reduction of GO, which can lead to high electrical conductivity and benefit the service performances of the composites.
The contents of SnO2 in SnO2@C@half-rGO and SnO2@C/rGO were measured to be 63.6 and 68.5 wt%, respectively (TGA, Supplementary Fig. S6). By the further analysis of the X-ray photoelectron spectroscopy (XPS) results, the mass fraction values of SnO2 in SnO2@C@half-rGO and SnO2@C/rGO were determined as 56.5 and 28.5 wt% (Supplementary Fig. S7, Supplementary Table S2), respectively, which are quite different from the TGA results (Supplementary Table S3). It is known that the photoelectron signal detected in XPS spectra is mostly restricted at the surface of the samples. The distinct different content values of SnO2 from TGA and XPS for SnO2@C/rGO (68.5 wt% to 28.5 wt%) is related to the wholly-wrapped structure (Supplementary Fig. S2), with SnO2 clusters covered with the dual thick carbon shell. On the contrary, the smaller difference analyzed by TGA and XPS (63.6 wt% to 56.5 wt%) for SnO2@C@half-rGO is ascribed to a thinner carbon shell, and thus gives another evidence for the formation of a partitioned-pomegranate-like structure.
The specific surface area (SSA) of SnO2@C@half-rGO is measured to be 375 m2 g−1 (Fig. 3c), which is more than twice that of LGO-based SnO2@C/rGO (177 m2 g−1) and three times that of the rGO-free comparison SnO2/C (124 m2 g−1). Compared with SnO2/C, the higher specific surface area of the GO-based samples can be interpreted as the dispersive effect of both SGO and LGO on SnO2 nanoparticles. However, the LGO sheets (SnO2@C/rGO) intend to wholly wrap the SnO2 clusters, and thus result in the less value than that of SnO2@C@half-rGO. The pore size is centered at ∼9 nm for all the three samples, derived from P123 during calcination process (Fig. 3d)27. The delicate nano-structure of SnO2@C@half-rGO along with the ultra-small size of the SnO2 nanoparticles is expected to afford remarkable lithium storage performance.
Galvanostatic charge−discharge tests were first conducted at the low current density of 100 mA g−1 (Fig. 4a). In contrast to the fast capacity fading of SnO2/C, only a slight declination was observed for SnO2@C@half-rGO and SnO2@C/rGO in the first few cycles with high capacities maintained. After 40 cycles, the discharge capacities of SnO2@C@half-rGO and SnO2@C/rGO were 879.6 mAh g−1 and 674.0 mAh g−1, respectively. Whereas, only 221.9 mAh g−1 was remained for the control sample SnO2/C. In order to find the reason why SnO2@C@half-rGO has a higher capacity than SnO2@C/rGO and SnO2/C, the cyclic voltammograms (CVs) of SnO2@C@half-rGO, SnO2@C/rGO and SnO2/C for the first three cycles were recorded (Supplementary Fig. S8). The alloying/dealloying reaction of LixSn species (corresponding to the anodic peaks at 0.1 V and cathodic peaks at 0.5 V) is similar for all the three samples. However, the conversion reactions of SnO2 and Sn are quite different. For SnO2@C@half-rGO, the anodic peak at 0.9 V (SnO2 converting into Sn) in the first sweep process is more apparent than that for SnO2@C/rGO and SnO2/C. There are two cathodic peaks (at 1.2 V and 1.8 V) for SnO2@C@half-rGO, indicating the reversible conversion of Sn back to SnO and further SnO2, and two corresponding anodic peak (at 0.9 V and 1.1 V) in the subsequent sweep process can be found. This reversible reaction has been deduced to be possible when the SnO2 particles are of nano-size28, and is contributive for the excess capacity of SnO2@C@half-rGO. For SnO2@C/rGO and SnO2/C, the weak and single peaks at 1.2 V (cathodic) and 0.9 V (anodic) reveal the less reversibility of the conversion reaction of SnO2 and Sn.

(a) Specific discharge capacity and coulombic efficiency of SnO2@C@half-rGO (red), SnO2@C/rGO (blue) and SnO2/C (grey) at 100 mA g−1. (b) Specific charge and discharge capacity of SnO2@C@half-rGO (red) and SnO2@C/rGO (blue) at 1 A g−1. (c) Mechanism of the structure evolution of SnO2@C@half-rGO and SnO2@C/rGO during lithiation/delithiation.
Interestingly, after tested for 200 cycles at 100 mA g−1, the discharge capacity of SnO2@C@half-rGO reached 1034.5 mAh g−1, while those of SnO2@C/rGO and SnO2/C reached 811.9 mAh g−1 and 296.3 mAh g−1, respectively. To determine the reason for this capacity recovery phenomenon, TEM image of SnO2@C@half-rGO after tests was taken as shown in Supplementary Fig. S9. Obviously, the porous cluster structure of SnO2 nanoparticles was preserved very well after 200 cycles. In coincidence with the up-to-date reported fundamental research of SnO2 anodes29, the SnO2 phase all changed to tetragonal Sn after 200 cycles, which is confirmed in SAED (Supplementary Fig. S9b inset) and XRD patterns (Supplementary Fig. S10). The Sn nanoparticles were observed to possess more separated and loose distribution on rGO sheets (Supplementary Fig. S9) than in the original morphology of SnO2@C@half-rGO (Fig. 2), which may be derived from the relaxation caused by repeated expansion-shrinkage process of Sn. Because of the fine particle size and the protective carbon shell, the Sn nanoparticles stay in adherent during the lithiation/delithiation reaction. Thus the incremental interface between Sn and amorphous carbon may provide more reaction sites for Li2O reversibly delithiating and lithiating, which contributes the increasing capacity29.
When tested at a high current density of 1 A g−1 (Fig. 4b), SnO2@C@half-rGO kept increasing for over 1000 cycles and reached 794.7 mAh g−1 at 1000 cycles. While the capacity of SnO2@C/rGO began to fade after 650 cycles and only maintained 431.8 mAh g−1 after 1000 cycles. The structure evolution mechanism of SnO2@C@half-rGO and SnO2@C/rGO during lithiation/delithiation is schematically shown in Fig. 4c. The wholly-wrapped structure in SnO2@C/rGO cannot bear cycling at such a high current density for hundreds of times, probably because the inadequate pore volume of the SnO2 clusters along with the severe volume change may have the rGO shell split. On the other hand, the half-supported structure of SnO2@C@half-rGO, like partitioned pomegranates, is not only beneficial to the swift Li+ transfer at high current density, but also capable to sustain the integrity of the rGO matrix for a long run.
The rate performance of SnO2@C@half-rGO and SnO2@C/rGO tested at stepped-incremental current densities is displayed in Fig. 5. In the first round of incremental current densities from 100 mA g−1 to 5 A g−1 throughout the first 70 cycles, the capacities of SnO2@C/rGO decreased much faster than those of SnO2@C@half-rGO. Only 179.7 mAh g−1 remained for SnO2@C/rGO at 5 A g−1 (the 70th cycle), while for SnO2@C@half-rGO at same condition 469.1 mAh g−1 remained. In the subsequent second round from the 71st to 140th cycle, the capacities of SnO2@C@half-rGO recovered to 821.1 mAh g−1 fast at 100 mA g−1 (the 72nd cycle), which was comparable to those at 100 mA g−1 in the first round (824.9 mAh g−1 for the 10th cycle). As for SnO2@C/rGO, the capacity of the 72nd cycle was only 534.0 mAh g−1, far smaller than that of the 10th cycle (665.0 mAh g−1), which means a big loss of lithium-storage capability after the preceding cycles. The weakness of the wholly-wrapped structure was evident, as the rGO-encapsulated structure of SnO2@C/rGO can seal off the mesopores inside the SnO2 clusters and thus is not beneficial to the rapid transfer of Li+, especially at high current densities. A long-period test at a high current density (5 A g−1) was carried out after the previous two round of rate test. The capacities of SnO2@C@half-rGO decreased to 370.3 mAh g−1 for the 10000th cycle, which is still comparable to the theoretical capacity of the conventional graphite anode for LIBs. Finally, the current density was tuned back to 100 mA g−1, and a gradual capacity recover process of SnO2@C@half-rGO was recorded. 972.3 mAh g−1 was reached in the final cycle, even higher than that of the 72nd cycle. This phenomenon can be ascribed to the activation effect of the very high current density30. It is worth mentioning that the high capacities, long service durability and satisfactory rate performance of SnO2@C@half-rGO are superior to most of reported SnO2-rGO composite anodes with hierarchical structures (shown in Supplementary Table S4).

Rate performance (discharge capacities) of SnO2@C@half-rGO (red) and SnO2@C/rGO (blue). The varying current is 100, 200, 500, 1000, 2000, 3000 and 5000 mA g−1 for 10 cycles each, following 100–3000 mA g−1 for 10 cycles each again, and 5 A g−1 for 9870 cycles until the final 100 mA g−1 for 10 cycles.
Encouraged by the excellent rate performance and the stability at high current densities of SnO2@C@half-rGO as anode for LIBs, we can reasonably expect its good performance as an anode for LIHCs. It is necessary to mention that SnO2 is essentially not a good candidate for LIHCs, because lithiation/delithiation of bulk SnO2 occurs in-depth rather than near-surface, which seems inconsistent with the demand for high power density of supercapacitors. However, as for SnO2@C@half-rGO here reported, the ultra-small size of the SnO2 nanoparticles, the ultimate conductivity of rGO, and the exposed mesopores among the SnO2 clusters have changed the intrinsic properties of SnO2 and make it a suitable anode for LIHCs.
Here, we chose the activated carbon with porous nature and graphene-like layers (gpC) as the cathode (Supplementary Fig. S11), which is prepared according to our previous report31. The prelithiated anodes were coupled with gpC cathode into complete LIHCs (SnO2@C@half-rGO//gpC or SnO2@C/rGO//gpC).
The CV curves of SnO2@C@half-rGO//gpC LIHC in different voltage windows of 0–3.0 V, 0–3.5 V, 0–3.8 V and 0–4.0 V all show a similar shape (Fig. 6a), indicating its capability to serve over the voltage window up to 4 V. It is acknowledged that with a same capacitance (C) value, the energy density (W) increases quadratically with the growth of voltage (V), according to the following formula:

(a) The CV curves of SnO2@C@half-rGO//gpC LIHC in different voltage windows of 0–3.0 V, 0–3.5 V, 0–3.8 V and 0–4.0 V at 50 mV s−1. The CV curves of (b) SnO2@C@half-rGO//gpC and (c) SnO2@C/rGO//gpC LIHC at different scan rates of 2, 5, 10, 20, 50 mV s−1 in the voltage windows of 0–4.0 V. Galvanostatic charge-discharge curves of (d) SnO2@C@half-rGO//gpC and (e) SnO2@C/rGO//gpC LIHC at different current densities of 0.1, 0.2, 0.5, 1.0, 2.0, 5.0, and 10.0 A g−1. (f) Specific capacitance values of SnO2@C@half-rGO//gpC (red) and SnO2@C/rGO//gpC (blue) LIHC calculated from the result shown in (d,e).
Therefore, the voltage window of 0–4 V was chosen so that high energy densities can be achieved. Different from the symmetric rectangular CV curves of EDLCs, the CV curves of SnO2@C@half-rGO//gpC LIHC (Fig. 6b) possess slightly asymmetric and humped shapes. The broad peaks between 2.5–4.0 V on both the anodic and cathodic curves (marked area) keep their presence with the scan rate increasing, which manifests that the faradaic pseudocapacitance of SnO2 in SnO2@C@half-rGO can be adequately exerted. In contrast, the control sample SnO2@C/rGO//gpC showed quite different properties (Fig. 6c). As the scan rate rises, the aforementioned broad peaks between 2.5–4.0 V gradually fade away. The peak-less shape of the CV indicates an EDLC-like property of SnO2@C/rGO. It is deduced that the wholly-wrapped structure of SnO2@C/rGO would impede the transfer of Li+ and hinder the capacitive performance from the SnO2 inside the rGO shell, especially in a fast charge−discharge process.
Figure 6d and e show the charge-discharge curves at different current densities for SnO2@C@half-rGO//gpC and SnO2@C/rGO//gpC LIHCs. Both the curves exhibit quasi-triangular shape. The specific capacitance values were calculated based on the sum mass of active materials on anodes and cathodes and are presented in Fig. 6 f. The capacitance of SnO2@C@half-rGO//gpC is higher than that of SnO2@C/rGO//gpC at each current density from 0.1–10.0 A g−1.
The energy density of SnO2@C@half-rGO//gpC at the average power density of ∼200 W kg−1 was determined to be 257 Wh kg−1. Even at a much higher average power density of ∼20 kW kg−1, SnO2@C@half-rGO//gpC can still maintain an energy density of 79 Wh kg−1. The values of the specific energy vs. specific power from this work are used to plot a Ragone chart (Fig. 7), in comparison with literature results of similar LIHC systems32,33,34,35,36,37,38. The dominant energy densities of both SnO2@C@half-rGO//gpC and SnO2@C/rGO//gpC over other LIHC systems at low power densities are attributed to the ultra-small size of the SnO2 nanoparticles and the mesopores between them, which create a large surface area for the sufficient redox reaction between SnO2 and Li+. Along with the increased power densities, the energy densities of SnO2@C@half-rGO//gpC maintain the superiority over other reported LIHC systems. It is easy to make out that at relatively high power densities there is more difference between the energy densities of SnO2@C@half-rGO//gpC and SnO2@C/rGO//gpC, which demonstrates the superiority of the half-open structure of SnO2@C@half-rGO. Although both SnO2@C@half-rGO//gpC and SnO2@C/rGO//gpC benefit from the mesopores inside the SnO2 clusters and the conductive rGO matrix outside, the half-open structure gives SnO2@C@half-rGO a larger exposed surface than SnO2@C/rGO for the high-speed transfer of Li+.

Ragone chart of SnO2@C@half-rGO//gpC and SnO2@C/rGO//gpC. The performance is compared with reported similar LIHC systems including Fe3O4/graphene//3D-graphene32, VN-RGO//AC33, porous NbN//AC34, MnO@graphene//HNC35, SnO2/Cu/CNT//AC36, Li4Ti5O12//activated bottom-up graphene37, and Li3V2(PO4)3-C//AC38.
Moreover, SnO2@C@half-rGO//gpC also held the coulombic efficiency nearly 100% throughout the 2000 cycles and exhibited excellent cycle stability (Fig. 8). The capacitance retention was up to 78.2% after 2000 cycles at 1.0 A g−1, much higher than the corresponding value of SnO2@C/rGO//gpC (56.5%, Supplementary Fig. S12).

The capacitance retention and coulombic efficiency of SnO2@C@half-rGO//gpC LIHC for 2000 cycles at 1.0 A g−1.
Our partitioned-pomegranate-structured materials are different from previously reported pomegranate-like particles that have a closed outer and inner surface. The partially opened outer surface of our material will ensure an unimpeded transfer of Li+ into the porous clusters, and at the same time the electrolyte-blocking amorphous carbon layer (like the pulp on pomegranate seeds) could prevent the SEI formation to the surface of the secondary particle. Owing to this structure, it is not surprised that the anode affords such a high cycling performance, especially high rate performance for LIBs, as well as superior performance as anodes for LIHCs.
Discussion
Inspired by nature, we designed a partitioned-pomegranate-like structure anode material which showed an enhanced lithium storage performance. The porous and micro-sized clusters of SnO2 nanoparticles are partially supported by small sheets of reduced graphene oxide. The mesopores inside the clusters remain open to electrolyte for the rapid transfer of Li+, which is superior to the wholly-wrapped structure originated from using large sheets of graphene oxide. The synthesized materials maintain a high reversible capacity (1034.5 mAh g−1 at 100 mA g−1 after 200 cycles), superior rate performance and excellent durability (370.3 mAh g−1 after 10000 cycles at 5 A g−1). When assembled as anode for LIHCs, a high energy density up to 257 Wh kg−1 at ∼200 W kg−1 was obtained. Even at a very high power density up to ∼20 kW kg−1, the LIHC can still afford 79 Wh kg−1. The result initiates a new architecture for graphene-based composite anode materials for LIBs and LIHCs, which is promising to be applied on other new-type anode materials. Furthermore, the performance of LIHCs reported herein is limited by the intrinsic electrochemical property of SnO2, and therefore, it is prospectively feasible to apply this graphene-half-supported structure to other materials such as MnO2 and RuO2, which are more suitable for pseudocapacitors.
Methods
Synthesis of SnO2 nanoparticles
The SnO2 nanoparticles were synthesized as colloidal dispersion. Typically, ammonia aqueous solution (25 wt%, 20 mL) was added dropwise into aqueous solution of SnCl4 under severe stirring. The precipitate was collected by centrifugation and washed with deionized water for several times until its pH was lower than 9.0. The washed precipitate was dispersed in deionized water (50 mL) again with sufficient stirring overnight, and the colloidal dispersion of SnO2 nanoparticles was thus obtained.
Synthesis of small sheets of graphene oxide (SGO)
The small sheets of graphene oxide was prepared by ultrasonically shredding the large sheets of graphene oxide (LGO), which was prepared in advance by modified Hummers method using flake graphite (325 mesh) as raw material39, 40. LGO aqueous dispension (2 mg mL−1, 500 mL) was tuned to alkalescent (pH = 10.0) and then treated by high-power ultrasonication for 2 h. Hydrochloric acid was dropped into the treated dispersion to lead to flocculation of the shredded graphene oxide, which was then collected by centrifugation and cleaned by dialysis. SGO was thus obtained and reserved as aqueous dispersion with concentration determined to be 5.0 mg mL−1.
Synthesis of SnO2@C@half-rGO
Pluronic P123 was introduced as surfactant to trigger the formation of SnO2 clusters12. Typically, 0.1 g of P123 was mixed with the as-prepared colloidal dispersion of SnO2 nanoparticles (40 mg mL−1, 3 mL), and after complete dissolution of P123, glucose (0.1 g, dissolved in 3 mL of deionized water) and the as-prepared SGO dispersion (4 mL) was added with sufficient stirring. After transferring the mixture into a polytetrafluoroethylene autoclave, kept at 180 °C for 3 h, the resultant sponge of SnO2-SGO was freeze-dried. The subsequent fumigation with hydrazine hydrate at 120 °C for 2 h41 and heat treatment at 500 °C for 2 h in nitrogen led to the reduction of SGO and the full carbonization of glucose derivatives, and SnO2@C@half-rGO was thus formed. For the synthesis of wholly-wrapped clusters of SnO2 nanoparticles (SnO2@C/rGO), same route were executed except LGO was utilized instead of SGO. Another control sample was prepared with the same route except that no GO was not introduced, and the product only contains SnO2 and amorphous carbon, which is simply named as SnO2/C.
Synthesis of graphitized porous carbon (gpC)
Rice husk was carbonized at 650 °C and then activated by KOH at 750 °C with a mass ratio of 1:1. The activated carbon was immersed in Fe(NO3)3 aqueous solution (0.2 mol L−1) with severe sonication treatment for 10 min. After drying at 80 °C overnight, the Fe(NO3)3/activated carbon mixture were graphitized at 800 °C for 3 h. The ferric impurity was washed by chlorhydric acid (5 wt%), and gpC was thus obtained.
Materials Characterization
XRD patterns were recorded on a Rigaku D/max2550VL/PC system, using Cu Kα radiation. SEM (JEOL JSM-6360LV, 15 kV), TEM and HRTEM (JEOL 2010, 200 kV) were used to observe the morphology of the samples. Nitrogen adsorption measurements were performed at 77 K on a Micromeritics ASAP 2020. BET specific surface area was calculated using the P/P 0 data between 0.10 and 0.25. Raman spectra were collected on a Renishaw in Via Raman microscope. TGA was conducted on a Perkin-Elmer TGA-7 Thermal Analyzer under O2 flow, with a heating rate of 20 °C min−1. XPS spactra were recorded on a Perkin-Elmer PHI-5400 spectrometer, using Mg Kα radiation as the excitation source.
Electrochemical Measurements
For half cell tests, all the samples (SnO2@C@half-rGO, SnO2@C/rGO and SnO2/C) were assembled into 2016 coin cells with Li0 foil as counter electrodes. To prepare working electrode, the samples were mixed with acetylene black and PVDF by a mass ratio of 8:1:1, and then coated on a copper foil with a mass/area ratio of ∼2 mg cm−2. Polypropylene membrane (Celgard 2500) was employed as separator. LiPF6 (1 mol L−1) in ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1, v/v) was used as electrolyte. All the cells were assembled in inert atmosphere and tested at room temperature. CV tests were done on a CHI-660D electrochemical workstation. Galvanostatic cycling tests were conducted on a LAND-CT2001A battery test system, with a voltage window of 0.01–3.00 V vs. Li/Li+. For LIHCs, the samples were first coated on copper foil and assembled in to coin cells with Li0 foil following the same process as above, and discharged to 0 V at a current density of 100 mA g−1. The prelithiated anodes were taken out and washed with DEC for three times. The gpC was mixed with acetylene black and PVDF by a mass ratio of 8:1:1, coated on aluminium foil to be cathode, and then assembled into coin cells with the prelithiated anodes. The mass ratio of cathode/anode in the LIHC devices was 3:1.
References
Fang, B., Kim, M.-S., Kim, J. H., Lim, S. & Yu, J.-S. Ordered Multimodal Porous Carbon with Hierarchical Nanostructure for High Li Storage Capacity and Good Cycling Performance. J. Mater. Chem. 20, 10253–10259 (2010).
Fang, B., Kim, J. H., Kim, M.-S. & Yu, J.-S. Hierarchical Nanostructured Carbons with Meso–Macroporosity: Design, Characterization, and Applications. Acc. Chem. Res. 46, 1397–1406 (2012).
Kim, M.-S. et al. Ultra-High Li Storage Capacity Achieved by Hollow Carbon Capsules with Hierarchical Nanoarchitecture. J. Mater. Chem. 21, 19362–19367 (2011).
Xing, Y., Wang, Y., Zhou, C., Zhang, S. & Fang, B. Simple Synthesis of Mesoporous Carbon Nanofibers with Hierarchical Nanostructure for Ultrahigh Lithium Storage. ACS Appl. Mater. Inter. 6, (2561–2567 (2014).
Cho, J. S., Park, J.-S. & Kang, Y. C. Preparation of Hollow Fe2O3 Nanorods and Nanospheres by Nanoscale Kirkendall Diffusion, and Their Electrochemical Properties for Use in Lithium-Ion Batteries. Sci. Rep. 6, 38933, doi:10.1038/srep38933 (2016).
Fan, L., Li, B., Rooney, D. W., Zhang, N. & Sun, K. In Situ Preparation of 3D Graphene Aerogels@Hierarchical Fe3O4 Nanoclusters as High Rate and Long Cycle Anode Materials for Lithium Ion Batteries. Chem. Commun. 51, 1597–1600 (2015).
Guo, C. X., Wang, M., Chen, T., Lou, X. W. & Li, C. M. A Hierarchically Nanostructured Composite of MnO2/Conjugated Polymer/Graphene for High-Performance Lithium Ion Batteries. Adv. Energy Mater. 1, 736–741 (2011).
Guan, Q. et al. Low Temperature Vacuum Synthesis of Triangular CoO Nanocrystal/Graphene Nanosheets Composites with Enhanced Lithium Storage Capacity. Sci. Rep 5, 10017, doi:10.1038/srep10017 (2015).
Fang, S., Shen, L., Zheng, H. & Zhang, X. Ge–Graphene–Carbon Nanotube Composite Anode for High Performance Lithium-Ion Batteries. J. Mater. Chem. A 3, 1498–1503 (2015).
Lin, D. et al. A High Tap Density Secondary Silicon Particle Anode Fabricated by Scalable Mechanical Pressing for Lithium-Ion Batteries. Energy Environ. Sci. 8, 2371–2376 (2015).
Zou, L. et al. Sn/C Non-Woven Film Prepared by Electrospinning as Anode Materials for Lithium Ion Batteries. J. Power Sources 195, 1216–1220 (2010).
Zhu, C. et al. Confined SnO2 Quantum-Dot Clusters in Graphene Sheets as High-Performance Anodes for Lithium-Ion Batteries. Sci. Rep. 6, 25829, doi:10.1038/srep25829 (2016).
Chan, C. K. et al. High-Performance Lithium Battery Anodes Using Silicon Nanowires. Nat. Nanotechnol. 3, 31–35 (2008).
Huang, J. Y. et al. In Situ Observation of the Electrochemical Lithiation of a Single SnO2 Nanowire Electrode. Science 330, 1515–1520 (2010).
Zhou, G. et al. Graphene-Wrapped Fe3O4 Anode Material with Improved Reversible Capacity and Cyclic Stability for Lithium Ion Batteries. Chem. Mater. 22, 5306–5313 (2010).
Loveridge, M. et al. Towards High Capacity Li-Ion Batteries Based on Silicon-Graphene Composite Anodes and Sub-Micron V-Doped LiFePO4 Cathodes. Sci. Rep 6, 37787, doi:10.1038/srep37787 (2016).
Chang, K. & Chen, W. L-Cysteine-Assisted Synthesis of Layered MoS2/Graphene Composites with Excellent Electrochemical Performances for Lithium Ion Batteries. ACS Nano 5, 4720–4728 (2011).
Cai, D. et al. Rational Synthesis of ZnMn2O4 Porous Spheres and Graphene Nanocomposite with Enhanced Performance for Lithium-Ion Batteries. J. Mater. Chem. A 3, 11430–11436 (2015).
Zhao, X., Hayner, C. M., Kung, M. C. & Kung, H. H. In-Plane Vacancy-Enabled High-Power Si−Graphene Composite Electrode for Lithium-Ion Batteries. Adv. Energy Mater 1, 1079–1084 (2011).
Xu, Y. et al. Holey Graphene Frameworks for Highly Efficient Capacitive Energy Storage. Nat. Commun. 5, 4554, doi:10.1038/ncomms5554 (2014).
Adhikari, M. P. et al. Nanoporous Activated Carbons Derived from Agro-Waste Corncob for Enhanced Electrochemical and Sensing Performance. Bull. Chem. Soc. Jpn 88, 1108–1115 (2015).
Liu, N. et al. A Pomegranate-Inspired Nanoscale Design for Large-Volume-Change Lithium Battery Anodes. Nat. Nanotechnol. 9, 187–192 (2014).
Akhavan, O., Ghaderi, E., Aghayee, S., Fereydooni, Y. & Talebi, A. The Use of a Glucose-Reduced Graphene Oxide Suspension for Photothermal Cancer Therapy. J. Mater. Chem. 22, 13773–13781 (2012).
Pan, H., Zhu, S. & Mao, L. Graphene Nanoarchitectonics: Approaching the Excellent Properties of Graphene from Microscale to Macroscale. J. Inorg. Organomet. Polymer. Mater. 25, 179–188 (2015).
Yang, D. et al. Chemical Analysis of Graphene Oxide Films after Heat and Chemical Treatments by X-Ray Photoelectron and Micro-Raman Spectroscopy. Carbon 47, 145–152 (2009).
Zhou, Y., Bao, Q., Tang, L. A. L., Zhong, Y. & Loh, K. P. Hydrothermal Dehydration for the “Green” Reduction of Exfoliated Graphene Oxide to Graphene and Demonstration of Tunable Optical Limiting Properties. Chem. Mater. 21, 2950–2956 (2009).
Soni, S., Brotons, G., Bellour, M., Narayanan, T. & Gibaud, A. Quantitative SAXS Analysis of the P123/Water/Ethanol Ternary Phase Diagram. J. Phys. Chem. B 110, 15157–15165 (2006).
Huang, B. et al. Novel Carbon-Encapsulated Porous SnO2 Anode for Lithium-Ion Batteries with Much Improved Cyclic Stability. Small 12, 1945–1955 (2016).
Lee, S.-Y. et al. Unveiling origin of additional capacity of SnO2 Anode in Lithium-Ion Batteries by Realistic Ex Situ TEM Analysis. Nano Energy 19, 234–245 (2016).
Sun, H. et al. High-Rate Lithiation-Induced Reactivation of Mesoporous Hollow Spheres for Long-Lived Lithium-Ion Batteries. Nat. Commun. 5, 4526, doi:10.1038/ncomms5526 (2014).
Li, Y. et al. Simple Fabrication of a Fe2O3/Carbon Composite for Use in a High-Performance Lithium Ion Battery. Carbon 52, 565–573 (2013).
Zhang, F. et al. A High-Performance Supercapacitor-Battery Hybrid Energy Storage Device Based on Graphene-Enhanced Electrode Materials with Ultrahigh Energy Density. Energy Environ. Sci. 6, 1623–1632 (2013).
Wang, R., Lang, J., Zhang, P., Lin, Z. & Yan, X. Fast and Large Lithium Storage in 3D Porous VN Nanowires–Graphene Composite as a Superior Anode Toward High-Performance Hybrid Supercapacitors. Adv. Funct. Mater. 25, 2270–2278 (2015).
Wang, P. et al. Porous Niobium Nitride as a Capacitive Anode Material for Advanced Li-Ion Hybrid Capacitors with Superior Cycling Stability. J. Mater. Chem. A 4, 9760–9766 (2016).
Yang, M. et al. Fabrication of High-Power Li-Ion Hybrid Supercapacitors by Enhancing the Exterior Surface Charge Storage. Adv. Energy Mater. 5 (2015).
Hsieh, C.-L., Tsai, D.-S., Chiang, W.-W. & Liu, Y.-H. A Composite Electrode of Tin Dioxide and Carbon Nanotubes and Its Role as Negative Electrode in Lithium Ion Hybrid Capacitor. Electrochim. Acta 209, 332–340 (2016).
Mhamane, D. et al. Silica-Assisted Bottom-Up Synthesis of Graphene-Like High Surface Area Carbon for Highly Efficient Ultracapacitor and Li-Ion Hybrid Capacitor Applications. J. Mater. Chem. A 4, 5578–5591 (2016).
Satish, R., Aravindan, V., Ling, W. C. & Madhavi, S. Carbon-Coated Li3V2(PO4)3 as Insertion Type Electrode for Lithium-Ion Hybrid Electrochemical Capacitors: An Evaluation of Anode and Cathodic Performance. J. Power Sources 281, 310–317 (2015).
Hummers, W. S. Jr & Offeman, R. E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 80, 1339–1339 (1958).
Sun, Z. et al. Synthesis of BiVO4@C Core–Shell Structure on Reduced Graphene Oxide with Enhanced Visible-Light Photocatalytic Activity. ChemSusChem 8, 2719–2726 (2015).
Zhou, X., Wan, L. J. & Guo, Y. G. Binding SnO2 Nanocrystals in Nitrogen-Doped Graphene Sheets as Anode Materials for Lithium-Ion Batteries. Adv. Mater. 25, 2152–2157 (2013).
Acknowledgements
The authors gratefully acknowledge the financial support from Ministry of Science and Technology of China (No. 2016YFA0202900), the National Science Foundation of China (NSFC, 51072117, 51672173), Guangdong Province (2016A010103018), Shanghai Science and Technology Committee (16520710900, 14JC1403300, 14520710100).
Author information
Authors and Affiliations
Contributions
C.Z., Z.C. and S.Z. proposed the study and designed the synthesis route. C.Z. performed the experiment. Y.L., H.P., X.M. and M.I. helped to optimize it. C.Z. analyzed experimental data, processed images, drew the schematic of the synthesis route and wrote the article. S.Z., Z.C., and D.Z. provided revisions to complete the article.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, C., Chen, Z., Zhu, S. et al. Construction of SnO2−Graphene Composite with Half-Supported Cluster Structure as Anode toward Superior Lithium Storage Properties. Sci Rep 7, 3276 (2017). https://doi.org/10.1038/s41598-017-03603-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03603-1
This article is cited by
-
Nitrogen-enriched graphene framework from a large-scale magnesiothermic conversion of CO2 with synergistic kinetics for high-power lithium-ion capacitors
NPG Asia Materials (2021)
-
Ultrathin MoS2 with expanded interlayers supported on hierarchical polypyrrole-derived amorphous N-doped carbon tubular structures for high-performance Li/Na-ion batteries
Nano Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
