
Abstract
Here we report de novo non-synonymous single-nucleotide variants (SNVs) by conducting whole exome sequencing of 18 trios consisting of Japanese patients with sporadic schizophrenia and their parents. Among nine SNVs, we explored the functional impact of the de novo mutation in TBL1XR1 [c.30 C > G (p.Phe10Leu)], a gene previously found to be associated with autism spectrum disorder and epilepsy. Protein structural analysis revealed that Phe10Leu mutation may decrease the structural stability of the TBL1XR1 protein. We demonstrate that Phe10Leu mutation alters the interaction of TBL1XR1 with N-CoR and β-catenin, which play critical roles in regulation of Wnt-mediated transcriptional activity. Consistently, TBL1XR1-mediated activation of Wnt signaling was up-regulated by Phe10Leu mutation. These results suggest that a de novo TBL1XR1 point mutation could alter Wnt/β-catenin signaling activity. Further studies are required to clarify the involvement of TBL1XR1 mutations in neuropsychiatric conditions.
Similar content being viewed by others


Introduction
Schizophrenia is a complex condition resulting from genetic and environmental etiological influences1. A meta-analysis of twin studies revealed that the point estimate of heritability of schizophrenia is 81%2. To date, multiple common and rare genetic variants have been identified as genetic risk factors for schizophrenia3,4,5, while there are a number of patients with no family history of schizophrenia, so called sporadic cases.
Lynch (2010) estimated that the average newborn acquires a total of 50–100 new mutations, resulting in approximately 0.86 novel amino acid-altering mutations per generation6. De novo mutations, such as single-nucleotide variants (SNVs), insertions and deletions (INDELs), and copy-number variants (CNVs) may contribute to the genetic etiology of sporadic schizophrenia and may explain the high prevalence rate of schizophrenia in general population. Recent trio-based studies using next-generation sequencing technology have identified de novo SNVs in sporadic schizophrenia7,8,9,10,11,12,13. Nonetheless, these rare SNVs are uncommon across studies, and molecular mechanisms of these mutations underlying schizophrenia still remain obscure. No whole-exome sequencing studies of sporadic schizophrenia have been previously reported in the Japanese population.
In the present study, we conducted whole exome sequencing of 18 trios consisting of patients with sporadic schizophrenia and their parents, and we identified de novo non-synonymous SNVs. We further examined the effect of the novel de novo TBL1XR1 mutation [c.30 C > G (p.Phe10Leu)] on the protein structure of TBL1XR1 and Wnt/β-catenin signaling pathway.
Results
De novo SNVs identified in schizophrenia trio samples by exome sequencing
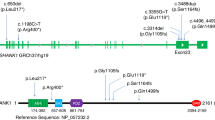
We conducted exome sequencing of 18 trios. On average, we obtained 15.4 GB of raw sequence data per sample, and 96.9% of these data were mapped to the reference genome (hg19). On detection of de novo mutations, we found 82 de novo SNVs in 18 trios. Of these 82 de novo SNVs, 17 were predicted to be non-synonymous mutations. Of these 17 de novo non-synonymous SNVs, we validated nine mutations in eight trios by Sanger sequencing (Table 1). Among these nine de novo non-synonymous SNVs, two mutations, one in the ABCD4 [ATP-binding cassette, sub-family D (ALD), member 4] gene and one in the TBL1XR1 [transducin (beta)-like 1 X-linked receptor 1] gene, were predicted as damaged by all of three software tools (Table 1). Given that de novo TBL1XR1 point mutations have been found in other neuropsychiatric conditions, including autism spectrum disorder (ASD) and West syndrome14, 15, we conducted subsequent structural and functional analyses of the observed de novo non-synonymous TBL1XR1 mutation [c.30 C > G (p.Phe10Leu)]. The TBL1XR1 point mutation (p.Phe10Leu) was not observed either in the independent 1,191 patients with schizophrenia nor in the 1,986 non-psychiatric control subjects.
Phe10Leu mutation impairs structural stability of TBL1XR1 protein
We next examined the effect of the TBL1XR1 point mutation (p.Phe10Leu) on the protein structure of TBL1XR1 (Fig. 1a). Surface areas of Phe and Leu amino acids were 175 and 137 Å2, respectively (Table 2a). Van der Waals (VdW) volumes of Phe and Leu were 135 and 124 Å2, respectively. Thus, we predict that substitution of Leu for Phe may decrease the volume and surface area of the 10th residue of TBL1XR1. TBL1XR1 is composed of two structural domains, the N-terminal domain (NTD) and tryptophan-aspartic acid 40 (WD40) repeat. The Phe10Leu mutation is located within the NTD. To assess the substitution in the context of protein structure, we built structural models of the NTD of the control and Phe10Leu TBL1XR1, and we obtained tetramer models of the NTD (Fig. 1b). In a monomer model of the control NTD, the side chain of Phe10 interacts with those of Tyr13, Arg14, Ile34, Ile39, and Val44 within the monomer (Fig. 1c). In the Phe10Leu model, the region around the 10th residue appears sparser than the control (Fig. 1d). We therefore calculated contact areas of the surrounding residues with the rest of the structures and found that those of the surrounding residues, except for Val44, were decreased in the Phe10Leu mutant (Table 2b). Calculated stability of protein structures was lower in the Phe10Leu NTD model than in the control (Table 2c). It is remarkable that statistics associated with VdW potentials were higher in the Phe10Leu NTD. Collectively, the results suggest that the Phe10Leu substitution of NTD decreases structural stability due to the decreases in the contact area of the 10th residue with the surrounding residues.

Structural models of TBL1XR1 N-terminal domain (NTD). (a) De novo mutation in TBL1XR1 [c.30 C > G (p.Phe10Leu)]. The chromatogram shows the mutation in the TBL1XR1 gene, which is observed in the proband (arrow) but not in the parents. (b) Overall structure of a homology model of tetrameric NTD of TBL1XR1. Monomers are depicted in distinct colors. (c) A monomer model of the control NTD is depicted as a ribbon model. Important residues are depicted as spheres. Gray, red and blue spheres indicate carbon, oxygen and nitrogen atoms, respectively, although all atoms of Phe10 are colored in cyan for clarity. (d) A monomer model of Phe10Leu NTD is depicted as a ribbon model. Leu10 is colored in cyan for clarity.
Phe10Leu mutation of TBL1XR1 alters Wnt signaling activity
TBL1XR1 is a component of the nuclear receptor corepressor (N-CoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) protein complex, which regulates transcriptional repression machinery16. TBL1XR1 also plays a critical role in recruiting β-catenin to Wnt target gene promoters for transcription activation17. To examine whether the Phe10Leu mutation affects TBL1XR1-mediated transcription mechanisms, either wild-type TBL1XR1 or the Phe10Leu mutant (TBL1XR1Phe10Leu) was exogenously expressed in 293T cells and co-immunoprecipitated with N-CoR and β-catenin. While the Phe10Leu mutation disturbed the interaction between TBL1XR1 and N-CoR, we observed increased binding of TBL1XR1 to β-catenin (Fig. 2a). Consistently, reduced binding of TBL1XR1 to N-CoR and increased interaction between TBL1XR1 and β-catenin were observed in HT22 cells, mouse hippocampal neuronal cells overexpressing TBL1XR1Phe10Leu (Fig. 2b). Next, we examined the effect of the Phe10Leu mutation on Wnt/β-catenin-mediated transcriptional activity by using the TOPFlash Wnt reporter assay18. Consistent with previous studies17, 19, overexpression of wild-type TBL1XR1 enhanced Wnt transcriptional activity, which was further up-regulated by Phe10Leu mutation (Fig. 2c). Interestingly, although Phe10Leu mutation decreases the interaction between TBL1XR1 and N-CoR, overexpression of N-CoR suppresses enhancement of Wnt transcriptional activity induced by either wild-type TBL1XR1 or Phe10Leu mutant (Fig. 2c).

The effect of the Phe10Leu mutation (F10L) on the protein interaction of TBL1XR1 with N-CoR and β-catenin as well as Wnt/β-catenin transcription activity. (a,b) Interaction of wild-type and F10L mutant TBL1XR1 (TBL1XR1Phe10Leu) with endogenous N-CoR and β-catenin was assessed in 293FT cells and HT22 hippocampal neuronal cells by co-immunoprecipitation experiments. TBL1XR1Phe10Leu displays stronger binding with β-catenin compared to wild-type TBL1XR1 (red arrowhead in top panel), while the binding of TBL1R1Phe10Leu and N-CoR is weaker than that of wild-type TBL1XR1 (red arrowhead in middle panel) (*P < 0.05 and **P < 0.01). The inputs of each protein are also shown (bottom panel). Full immunoblots are presented in Supplementary Figure. (c) The TOPFlash Wnt reporter assay showed that overexpression of wild-type TBL1XR1 increased Wnt transcription activity, which was further enhanced by an F10L mutation in TBL1XR1 (*P < 0.05 and **P < 0.01). Overexpression of N-CoR suppresses an increase in Wnt transcriptional activity induced by either wild-type TBL1XR1 or F10L mutant (**P < 0.01). Luciferase activities were determined 48 hours post-transfection and normalized against Renilla values. Bars represent averages of each group in three independent experiments. AU, arbitrary unit. All data are presented as the mean ± s.e.m.
Discussion
To the best of our knowledge, this is the first study conducting trio-based exome sequencing using Japanese subjects with sporadic schizophrenia. The observed exome point mutation rate in schizophrenia in the present study was similar to those of previous exome sequencing studies11, 13. While no genes reported in previous trio-based genetic studies of schizophrenia7,8,9,10,11,12,13 was detected in our cohort, we identified the de novo mutation in TBL1XR1 (p.Phe10Leu), a gene previously found to be associated with neuropsychiatric conditions. O’Roak et al. reported two de novo point mutations (p.Leu282Pro and p.Ile397SerfX19) in sporadic cases of ASD14, 20. Saitsu and colleagues found a de novo point mutation (p.Gly70Asp) in a patient with West syndrome and three missense variants in this gene (p.Ala116Ser, p.Gly405Glu, and p.Asn407Ser) in patients with epilepsy15. Deletions on 3q26.32, encompassing the TBL1XR1 gene, have been reproducibly associated with intellectual disability21, 22. These results suggest that mutation of the TBL1XR1 gene may contribute to a genetic vulnerability to multiple neurodevelopmental psychiatric conditions.
The results we obtained in protein structural and functional analysis suggest potential pathogenic impact of Phe10Leu mutation in TBL1XR1 function. TBL1XR1 is a component of the quaternary corepressor complex composed of N-CoR, SMRT and histone deacetylase 3 (HDAC3), which plays a key role in regulating transcription repression16, 23,24,25. Recent studies demonstrated that TBL1XR1 also plays a critical role in recruiting β-catenin to Wnt target gene promoters for transcriptional activation17. These results suggest that TBL1XR1 may act as a molecular switch for transcriptional activation and repression, which may be regulated by its posttranslational modification, such as SUMOylation26. The results of our protein structural analysis indicate that Phe10Leu substitution in the NTD of TBL1XR1 decreases structural stability of the NTD, which may influence binding to other components of the corepressor complex. In fact, we observed that Phe10Leu substitution decreases interaction between TBL1XR1 and N-CoR, whereas binding of TBL1XR1 with β-catenin is increased, which may explain observed up-regulation of Wnt/β-catenin-mediated transcriptional activity induced by Phe10Leu substitution.
Accumulating evidences suggest that altered Wnt signaling may be implicated in etiopathophysiologies of neurodevelopmental psychiatric conditions, such as schizophrenia and ASD14, 27, 28. Up-regulation of Wnt/β-catenin signaling and dysregulated function of NCoR have been documented in multiple types of cancers29, 30. High expression of TBL1XR1 has been reported to be associated with a poor prognosis of colorectal cancer31. Consistently, Wnt/β-catenin signaling and the N-CoR/TBL1XR1 complex play critical roles in cell proliferation and differentiation17, 26, 29. Thus, it would be of interest to investigate how altered Wnt signaling pathway induced by TBL1XR1 mutations may affect cellular processes in brain development.
The ABCD4 point mutation (p.lle344Thr) found in the present study was also predicted as damaged by in silico functional analysis. ABCD4 is an ABC transporter that has been classified as a member of the D subfamily of peroxisomal ABC transporters, and mutations in ABCD4 cause a new inborn error of vitamin B12 (cobalamin) metabolism32. Given that decreased levels of cobalamin have been reported in the frontal cortex in schizophrenia and autism compared with controls33, the effect of ABCD4 point mutations in the metabolic processes of cobalamin in brains should be investigated in future studies.
In conclusion, we report nine novel de novo non-synonymous SNVs as a result of the whole-exome sequencing of 18 trios. In particular, a de novo TBL1XR1 point mutation could alter Wnt/β-catenin signaling activity, supporting the potential involvement of altered Wnt signaling pathway in neurodevelopmental psychiatric disorders. Further studies are required to clarify the involvement of TBL1XR1 mutations in neuropsychiatric conditions.
Materials and Methods
Sample collection
We recruited 18 trios of patients with schizophrenia and their unaffected parents from the Tokushima University Hospital, Ehime University Hospital, Kochi University Hospital, and Nagasaki University Hospital in Japan. Patients with schizophrenia (1191 in total, 696 men and 495 women, mean age: 59.5 ± 14.5 years) were independently recruited from Tokushima University Hospital in Japan. Schizophrenia was diagnosed according to Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria by at least two expert psychiatrists based on extensive clinical interviews and a review of medical records (Supplementary Table 1). Non-psychiatric control subjects (1986 in total, 833 men and 1153 women, mean age: 38.6 ± 13.3 years) were selected from volunteers recruited from hospital staff, students, and company employees with no documented history of mental illness or psychiatric problems. All subjects were of Japanese origin. This study was approved by the ethics committees of Ehime, Kochi, Nagasaki, and Tokushima Universities. All enrolled participants provided their signed written informed consent for participation. This study was carried out in accordance with the World Medical Association’s Declaration of Helsinki.
Exome capture and sequencing
Genomic DNA was extracted from blood leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Exome enrichment was performed by the TruSeq DNA Sample Prep Kits and TruSeq Exome Enrichment Kit (Illumina, San Diego, CA, USA). Exome sequencing was performed on a HiSeq1000/1500 (Illumina, San Diego, CA, USA).
Data processing for identification of de novo SNVs
Raw sequencing data for each individual was mapped to the human reference genome (build hg19) by using the Burrows-Wheeler Aligner (BWA v0.5.9)34. BWA-generated SAM files were converted into BAM format, sorted and indexed using SAMtools v.0.1.1935, then processed by Picard (v1.90) to mark duplicated reads. The BAM-formatted files were further processed using the Genome Analysis Toolkit (GATK, v2.6-4 or v2.6-5) according to the GATK’s best-practice recommendations. In brief, the BAM files were processed with GATK tools (RealignerTargetCreator, IndelRealigner, BaseRecalibrator and PrintReads) to perform local realignment around indels and recalibration of the base quality scores, followed by the data compression with the downstream GATK tool (ReduceReads). When using all of the processed BAM files from 18 trios, multi-sample variant calling with the UnifiedGenotyper tool in GATK (v2.6-5) was done to identify SNV and indel candidates. The resulting Variant Call Format file (VCF, version 4.1) was applied to variant quality score recalibration with the GATK VariantRecalibrator tool. Genomic annotations associated with the variants in the VCF file were added using snpEff (v2.0.5d) with the GRCh37.64 database. According to the GATK’s recommendations for variant filtering, the annotated VCF file was used to extract potential true positive variants on the following values: QD < 2.0, MQ < 40.0, FS > 60.0, HaplotypeScore > 13.0, MQRankSum < −12.5, and ReadPosRankSum < −8.0. After the multi-sample VCF file was split into each of the 18 trios using GATK, the trio-based VCF files generated were used to obtain variants that violated Mendel’s law of segregation in the respective families as candidate de novo variants. Of these variants, we extracted de novo SNV candidates on the following settings: 1) read depth of coverage at SNV sites were 30 or more in both the proband and parents from a trio of interest, 2) SNVs were not present in the dbSNP v137 database and 3) SNVs were seen in only the proband of interest. Candidate de novo non-synonymous SNVs were validated by standard Sanger sequencing on an ABI 3130xl DNA Analyzer. Genotyping of the TBL1XR1 point mutation [c.30 C > G (p.Phe10Leu)] was performed using a commercially available TaqMan probe with the Applied Biosystems 7500 Fast Real Time PCR System, according to the protocol recommended by the manufacturer (Applied Biosystems, CA, USA).
In silico functional analysis
To predict the effect of de novo non-synonymous SNVs on protein function, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)36, SIFT (http://sift.jcvi.org/)37, and PROVEAN (http://provean.jcvi.org/index.php)38 were used.
Protein structural analysis
Structural models of the N-terminal domain (NTD) of TBL1XR1 were built with homology modeling method by using the Swiss Model39. The 2xtc.pdb template was used for building the models. Models contained residues 2–75 of TBL1XR1. Folding energies of the models were calculated with FoldX40. Contact area for each residue was calculated by using PDBePISA41. Structural illustrations were depicted with Swiss PDB Viewer42.
Plasmids and antibodies
The FLAG-tagged TBL1XR1 expression construct was a gift from Dr. Cun-Yu Wang (University of California, Los Angeles)17. The FLAG-tagged mutant TBL1XR1 (TBL1XR1Phe10Leu) expression construct was made by a PCR-based mutagenesis protocol43. For biochemical experiments, the following antibodies were used: rabbit polyclonal anti-β-catenin antibody (Sigma, Beverly, MA, USA), rabbit polyclonal anti-N-CoR antibody (EMD Millipore, Billerica, MA, USA), as well as mouse monoclonal and rabbit polyclonal anti-FLAG antibodies (Sigma, Beverly, MA, USA).
Transfection and Wnt/β-catenin activity assays with luciferase reporter system
Human embryonic kidney 293FT cells were maintained in Dulbecco’s modified Eagle’s medium nutrient mixture F-12 (DMEM/F12 (1:1); Gibco BRL, Gaithersburg, MD, USA) containing 10% FBS at 37 °C in a 5% CO2/95% air atmosphere. Twelve-well plates were seeded with 1 × 106 293FT cells in a medium containing 1% FBS. Transfection of the TOPFlash reporter plasmid, Renilla luciferase cDNA in an SV40 (pRL-SV40, as an internal control) and N-CoR expression construct, together with FLAG-tagged TBL1XR1, TBL1XR1Phe10Leu or an empty (mock) vector, was carried out with Lipofectamine 2000 (Invitrogen, Waltham, MA, USA). Two days after transfection, cells were lysed, and Wnt/β-catenin activity was measured using the Promega Dual Luciferase Reporter Assay System (Promega, Fitchburg, WI, USA) and a FLUOstar Luminometer (BMG Labtech, Ortenberg, Germany).
Co-immunoprecipitation
293FT cells and HT22 cells transfected with the FLAG-tagged TBL1XR1 or TBL1XR1Phe10Leu expression construct or mock vector were lysed in IP buffers [50 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 1% NP-40, 0.3% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and protease inhibitor mixture (Roche, Basel, Switzerland)] and [50 mM Tris·HCl, pH 7.4, 150 mM sodium chloride, 0.1% NP-40, and protease inhibitor mixture], respectively. Supernatant fractions obtained after centrifugation at 12,000 g for 15 minutes were incubated with primary antibodies and protein G Plus/Protein A agarose (Calbiochem, Darmstadt, Germany). Immunoprecipitates were analyzed with SDS-PAGE followed by Western blotting after extensive washing. Endogenous β-catenin and N-CoR binding to exogenous TBL1XR1 and TBL1XR1 Phe10Leu was analyzed by densitometry.
References
Sullivan, P. F. The genetics of schizophrenia. PLoS Med. 2, e212, doi:10.1371/journal.pmed.0020212 (2005).
Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192, doi:10.1001/archpsyc.60.12.1187 (2003).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Levinson, D. F. et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168, 302–316, doi:10.1176/appi.ajp.2010.10060876 (2011).
McClellan, J. & King, M.-C. Genomic analysis of mental illness: a changing landscape. JAMA 303, 2523–2524, doi:10.1001/jama.2010.869 (2010).
Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. 107, 961–968, doi:10.1073/pnas.0912629107 (2010).
Girard, S. L. et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863, doi:10.1038/ng.886 (2011).
Xu, B. et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868, doi:10.1038/ng.902 (2011).
Xu, B. et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369, doi:10.1038/ng.2446 (2012).
Gulsuner, S. et al. Spatial and Temporal Mapping of De Novo Mutations in Schizophrenia to a Fetal Prefrontal Cortical Network. Cell 154, 518–529, doi:10.1016/j.cell.2013.06.049 (2013).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184, doi:10.1038/nature12929 (2014).
McCarthy, S. E. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658, doi:10.1038/mp.2014.29 (2014).
Guipponi, M. et al. Exome Sequencing in 53 Sporadic Cases of Schizophrenia Identifies 18 Putative Candidate Genes. PLoS ONE 9, e112745, doi:10.1371/journal.pone.0112745 (2014).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250, doi:10.1038/nature10989 (2012).
Saitsu, H. et al. A girl with West syndrome and autistic features harboring a de novo TBL1XR1 mutation. J. Hum. Genet. 59, 581–583, doi:10.1038/jhg.2014.71 (2014).
Oberoi, J. et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18, 177–184, doi:10.1038/nsmb.1983 (2011).
Li, J. & Wang, C.-Y. TBL1–TBLR1 and β-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat. Cell Biol. 10, 160–169, doi:10.1038/ncb1684 (2008).
Ishizuka, K. et al. DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473, 92–96, doi:10.1038/nature09859 (2011).
Ogawa, S. et al. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA. 101, 14461–14466, doi:10.1073/pnas.0405786101 (2004).
O’Roak, B. J. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622, doi:10.1126/science.1227764 (2012).
Pons, L. et al. A new syndrome of intellectual disability with dysmorphism due to TBL1XR1 deletion. Am. J. Med. Genet. A. 167A, 164–168, doi:10.1002/ajmg.a.v167.1 (2015).
Tabet, A.-C. et al. De novo deletion of TBL1XR1 in a child with non-specific developmental delay supports its implication in intellectual disability. Am. J. Med. Genet. A. 164A, 2335–2337, doi:10.1002/ajmg.a.36619 (2014).
Guenther, M. G. et al. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 14, 1048–1057 (2000).
Guenther, M. G., Barak, O. & Lazar, M. A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 21, 6091–6101, doi:10.1128/MCB.21.18.6091-6101.2001 (2001).
Yoon, H.-G. et al. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 22, 1336–1346, doi:10.1093/emboj/cdg120 (2003).
Choi, H.-K. et al. Reversible SUMOylation of TBL1-TBLR1 regulates β-catenin-mediated Wnt signaling. Mol. Cell 43, 203–216, doi:10.1016/j.molcel.2011.05.027 (2011).
De Ferrari, G. V. & Moon, R. T. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553, doi:10.1038/sj.onc.1210064 (2006).
Topol, A. et al. Altered WNT Signaling in Human Induced Pluripotent Stem Cell Neural Progenitor Cells Derived from Four Schizophrenia Patients. Biol. Psychiatry 78, e29–e34, doi:10.1016/j.biopsych.2014.12.028 (2015).
Wong, M. M., Guo, C. & Zhang, J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2, 169–187 (2014).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205, doi:10.1016/j.cell.2012.05.012 (2012).
Liu, H. et al. Correlations between TBL1XR1 and recurrence of colorectal cancer. Sci. Rep. 7, 44275, doi:10.1038/srep44275 (2017).
Coelho, D. et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 44, 1152–1155, doi:10.1038/ng.2386 (2012).
Zhang, Y. et al. Decreased Brain Levels of Vitamin B12 in Aging, Autism and Schizophrenia. PloS One 11, e0146797, doi:10.1371/journal.pone.0146797 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25, 1754–1760, doi:10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079, doi:10.1093/bioinformatics/btp352 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249, doi:10.1038/nmeth0410-248 (2010).
Sim, N.-L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–457, doi:10.1093/nar/gks539 (2012).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PloS One 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–258, doi:10.1093/nar/gku340 (2014).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–388, doi:10.1093/nar/gki387 (2005).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797, doi:10.1016/j.jmb.2007.05.022 (2007).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723, doi:10.1002/elps.1150181505 (1997).
Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59, doi:10.1016/0378-1119(89)90358-2 (1989).
Acknowledgements
The authors appreciate all the volunteers who understood our study purpose and participated in this study, as well as the physicians who helped collect clinical data and blood samples at the mental hospitals. The authors would also like to thank Mrs. Akemi Okada for her technical assistance and Ms. Nada Rendradjaja for critical reading of the manuscript. This work was supported in part by Japan Science and Technology Agency, CREST and a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (T.O.), the Uehara Memorial Foundation (S.N.), R01 MH-091230 (A.K.), R01 DA041208 (A.K.), P50 MH-094268 (A.K.), R21 AT008547 (A.K.), and JHU Catalyst Award (A.K.).
Author information
Authors and Affiliations
Contributions
Numata S. designed the study. Kamiya A., Numata S., and Ohmori T. managed the research. Ito K., Kinoshita M., Nishi A., Numata S., Saito A., Tajima A., and Zhu X. performed experiments. Kato Y. and Fukui K. carried out protein structural analysis. Imoto I., Nishi A., Tajima A. undertook the statistical analysis. Imamura A., Kinoshita M., Kurotaki N., Numata S., Ochi S., Ono S., Shimodera S., and Ueno S. collected samples. Imoto I., Iwata N., Ohmori T., and Tajima A. helped to interpret data and edited the manuscript. Nishi A., Kamiya A., Kato Y., and Numata S. wrote the manuscript. All authors contributed to and have approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishi, A., Numata, S., Tajima, A. et al. De novo non-synonymous TBL1XR1 mutation alters Wnt signaling activity. Sci Rep 7, 2887 (2017). https://doi.org/10.1038/s41598-017-02792-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02792-z
This article is cited by
-
The spectrum of neurological presentation in individuals affected by TBL1XR1 gene defects
Orphanet Journal of Rare Diseases (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
