
Abstract
The limitation of 16S rRNA gene sequencing (DNA-based) for microbial community analyses in water is the inability to differentiate live (dormant cells as well as growing or non-growing metabolically active cells) and dead cells, which can lead to false positive results in the absence of live microbes. Propidium-monoazide (PMA) has been used to selectively remove DNA from dead cells during downstream sequencing process. In comparison, 16S rRNA sequencing (RNA-based) can target live microbial cells in water as both dormant and metabolically active cells produce rRNA. The objective of this study was to compare the efficiency and sensitivity of DNA-based, PMA-based and RNA-based 16S rRNA Illumina sequencing methodologies for live bacteria detection in water samples experimentally spiked with different combination of bacteria (2 gram-negative and 2 gram-positive/acid fast species either all live, all dead, or combinations of live and dead species) or obtained from different sources (First Nation community drinking water; city of Winnipeg tap water; water from Red River, Manitoba, Canada). The RNA-based method, while was superior for detection of live bacterial cells still identified a number of 16S rRNA targets in samples spiked with dead cells. In environmental water samples, the DNA- and PMA-based approaches perhaps overestimated the richness of microbial community compared to RNA-based method. Our results suggest that the RNA-based sequencing was superior to DNA- and PMA-based methods in detecting live bacterial cells in water.
Similar content being viewed by others


Introduction
Water-borne pathogens and their associated diseases pose a high risk to public health1. The majority of research in this area initially focused on fecal-associated pathogens, but in recent years the research has broadened considerably towards non-fecal opportunistic pathogens, such as several species within genera Legionella and Mycobacteria2, 3. Culture-based methods have long been used to detect microbial pathogens in the environment; however, these methodologies are still labor intensive, lengthy, and are not able to detect difficult to cultivate species. As such, only less than 1% of the microorganisms present in the environment, such as in natural water and soil samples, have been cultured with the currently known methods4. Although development of culturomic techniques in recent years has helped to culture not previously cultivated organisms that are corresponding to a sequence(s) obtained from a mixed microbial community using 16S rRNA or shotgun whole genome sequencing approaches5, the methodology is yet far from being perfect, and thus, sequencing approaches are still the most reliable tool for characterization of members of a microbial community. That being said, DNA-based identification methods do not discriminate between DNA from live (dormant cells as well as growing or non-growing metabolically active cells) and dead microbial cells. This becomes problematic as DNA of dead bacterial cells can persist in the environment depending on the environmental conditions6. For instance, DNA of dead bacterial cells has been shown to persist for 25 days in stream water7 and 70 days in soil8. Considering the slow decay rate of DNA from dead bacterial cells, the DNA-based detection methods have a tendency to overestimate bacterial richness and abundance in a sample; hence, leading to false positive results of live pathogens making them less suitable tool for water quality check.
Specialized methods to detect live bacterial cells are becoming popular. This includes the use of propidium monoazide (PMA), a DNA-intercalating agent that only penetrates membrane-compromised/dead bacterial cells and forms photo-induced crosslinks after exposure to light to allow for the selective removal of dead cells from downstream DNA applications9,10,11. The PMA in combination with real-time quantitative PCR (qPCR) analysis has been used to inhibit amplification of both extracellular DNA and DNA in dead or membrane-compromised bacterial cells12. Gensberger et al.13 evaluated DNA-qPCR and PMA-qPCR assays for evaluating microbial water quality. These authors reported that PMA-qPCR compared to DNA-qPCR resulted in higher sensitivity and specificity in detecting Escherichia coli, Enterococcus spp. and Pseudomonas aeruginosa, as well as coliforms (Enterobacteriaceae family), which are indictor species/groups for water microbial quality control.
Several recent studies10, 11, 14 have combined high-throughput sequencing and PMA-based methodologies to detect live cells in human and environmental samples. Despite its advantages, the PMA-based approach has known practical and theoretical limitations. For example, the incubation temperature and duration, as well as the concentration of PMA used need to be optimized based on the levels of suspended solids and microbial biomass in the water. These optimizations are based on trial and error, which is costly and time consuming to determine the correct combination of all the abovementioned factors in order to generate reproducible, sensitive and accurate data15. Additionally, the technique is also known to lead to false-positive signals due to the penetration of PMA via damaged cell membranes of live microorganisms12, 15.
Compared to DNA, RNA degrades more rapidly in the environment16, 17. The estimated turnover time or half-lives of prokaryotic RNA is about few minutes. For example the half-lives of E. coli RNA is around 5 min and that of Bacillus subtilis ranges from 7 to 15 min18, 19. Thus, RNA might be a more suitable target for studying live members of water microbial community. To our knowledge there has been no comparison between DNA-, PMA-, and RNA-based 16S rRNA sequencing for detection of live bacterial cells in water samples. The objective of this study was to compare these three techniques for detection of live bacteria in water samples experimentally spiked with different combination of known bacteria or obtained from different sources. Water source characteristics included ultra-pure HPLC-grade water spiked with gram-positive, gram-negative and acid-fast bacterial strains; treated drinking water that showed negative or positive for total coliforms; as well as lake and river water that showed positive for total coliforms and had relatively small and large sediment load, respectively.
Results and Discussion
Taxonomic classification of clustered OTUs in mini-microbial communities of spiked water samples determined using DNA-, PMA- and RNA-based methods are presented in Figs 1, 2 and 3. We also further used PCoA and PERMANOVA analyses to visualize and compare the variations in β-diversity among mini-microbial communities (Fig. 4; Supplementary Table S1).

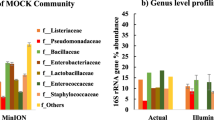
OTU compositions of mini-microbial communities in spiked water samples consisting of (a) all live cells of Bacillus amyloliquefaciens, Mycobacterium smegmatis, Escherichia coli and Yersinia enterocolitica; (b) all dead cells of the aforementioned species; (c) dead cells of B. amyloliquefaciens and M. smegmatis, and live cells of E. coli and Y. enterocolitica; (d) live cells of B. amyloliquefaciens and M. smegmatis, and dead cells of E. coli and Y. enterocolitica; determined by the DNA-, RNA-, and PMA-based 16S rRNA MiSeq Illumina sequencing.

Average sequences in mini-microbial communities of spiked water consisting of (a) all live cells of Bacillus amyloliquefaciens, Mycobacterium smegmatis, Escherichia coli and Yersinia enterocolitica; (b) all dead cells of the aforementioned species; (c) dead cells of B. amyloliquefaciens and M. smegmatis, and live cells of E. coli and Y. enterocolitica; (d) live cells of B. amyloliquefaciens and M. smegmatis, and dead cells of E. coli and Y. enterocolitica; determined using DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing. *P < 0.05.

Compositions of mini-microbial communities of spiked water consisting of (a) all live cells of Bacillus amyloliquefaciens, Mycobacterium smegmatis, Escherichia coli and Yersinia enterocolitica; (b) all dead cells of the aforementioned species; (c) dead cells of B. amyloliquefaciens and M. smegmatis, and live cells of E. coli and Y. enterocolitica; (d) live cells of B. amyloliquefaciens and M. smegmatis, and dead cells of E. coli and Y. enterocolitica; determined using DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing.

Principal coordinates analysis (PCoA) of unweighted UniFrac distances comparing the β-diversity of mini-microbial communities of spiked water among DNA-, PMA-, RNA-based methods. The mini-microbial communities compositions were as follow: (a) all live cells of Bacillus amyloliquefaciens, Mycobacterium smegmatis, Escherichia coli and Yersinia enterocolitica; (b) all dead cells of the aforementioned species; (c) dead cells of B. amyloliquefaciens and M. smegmatis, and live cells of E. coli and Y. enterocolitica; (d) live cells of B. amyloliquefaciens and M. smegmatis, and dead cells of E. coli and Y. enterocolitica; determined using DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing.
Each of the three methods produced results that were different from the original mixing proportions of all live, all dead, or live and dead combinations of bacteria (Fig. 3). In our mixture design experiment, similar to this type of study20, the assumption was that the proportion of sequenced OTUs should only depend on the relative proportion of spiked cells into the mixture and the differences between the DNA-, PMA- and RNA-based methodologies, such as the efficiency of DNA vs. RNA extraction, the effect of PMA on the DNA extraction outcome, and the efficiency of RNA to cDNA conversion. However, this assumption was not completely correct as additional bias can be introduced due to other factors. Brooks et al.21 conducted a set of mixture design experiments during which seven spiked species were either equally mixed first followed by DNA extraction, PCR amplification and high-throughput sequencing, or separately DNA extracted and the DNA was equally mixed for PCR amplification followed by sequencing, or DNA was separately extracted and PCR was separately performed and the PCR products were equally mixed for high-throughput sequencing. The authors reported that mini-microbial community compositions varied among the three experiments with highest similarity to the actual mixing proportions of cells when DNA extractions and PCR amplifications were performed separately on each individual bacterium. The authors concluded that the level of bias was highly dependent on the bacterial species in the mix. In other words, a bacterium’s proportion in the mini-microbial community could be overestimated or understimated due to presence of other bacteria. The study indicated that among factors contributing to such bias perhaps cell lysability, gram-negativity, GC content, and differential amplification efficiency of primers for each species were the major contributing factors. The differences in the genome content of species or in their copy numbers of 16S rRNA genes had less impact on the observed bias than the aforementioned factors.
In this context, Fig. 2 indicates that the total sequence numbers obtained from DNA-, PMA- and RNA-based methods were in closer range when all live (19,210 to 24,801), or a mixture of live and dead cells (19,597 to 29,753) were spiked into the samples. In contrast, when all dead cells were spiked, the number of sequences was lower (P < 0.001) in the RNA-based method (8,659) compared to DNA- (23,875) and PMA-based (29,753) methods. The high number of sequences detected by DNA- and PMA-based methods when all dead cells were spiked indicates that these methodologies are not efficient for differentiating between live and dead cells. The RNA-based method, which involved the extraction of microbial cellular RNA from metabolically active cells following by its conversion to cDNA and sequencing, while was superior for detection of live bacterial cells still identified a considerable number of 16S rRNA targets from samples spiked with dead cells (Fig. 3b). Figure 3b clearly shows that the majority (68%) of detected 16S rRNA sequences using RNA-based method belonged to M. smegmatis, which similar to other mycobacteria has a cell wall structure that is more rigid than other bacterial species22. As a result, we speculate that detected 16S rRNA targets using RNA-based method (Fig. 3b) was perhaps due to inefficiency of heat treatment step for complete breakdown and inactivation of M. smegmatis cells leaving a proportion of cells to be metabolically active. The DNA- and PMA-based analyses of these samples further supports this hypothesis (Fig. 3b, Supplementary Table S1). Using DNA-based approach all 4 dead spiked species were identified in the community with the lowest proportion for Mycobacterium (18%) and the highest for Escherichia (35%) suggesting that when cells were properly disrupted (heat exposure followed by chemical and mechanical lysis during the extraction procedures) and DNA is exposed, DNA-based methods can amplify the DNA irrespective of its source (live or dead cells). In contrast, when PMA-based method was employed to assess the mini-microbial community of spiked water with all dead cells, Escherichia, Bacillus, and Yersinia were highly amplified ranging from 22% to 44% of the sequences, while Mycobacterial proportion was only 1%. The results firstly indicated that PMA had low efficiency for removal of DNA from dead cells when microbial biomass was high, and secondly suggested that a large proportion of Mycobacterial cells – compared to other species in the mix – were perhaps still intact, which prevented PMA from penetration and binding to DNA.
Our results are supported with previous observations showing that PMA treated DNA from dead gram-negative E. coli cells were still detectable by qPCR as the DNA from dead/membrane-compromised cells were not completely removed by PMA treatment23. Authors articulated that the levels of biomass in water samples may interfere with the ability of the PMA-qPCR method to detect live cells. Varma et al.12 similarly showed that both gram-positive and gram-negative bacterial DNA present in wastewater can be simultaneously detected by qPCR after treatment with PMA when the levels of biomass were high.
Comparison of the prescribed proportions of bacteria with the results of the DNA-based sequencing method when all live bacteria were spiked can be used to evaluate whether the observed proportions of bacteria were overestimated or underestimated by the presence of other bacteria in the mini-microbial community (Fig. 3a). Our results showed that the sequence reads obtained from mini-microbial community were dominated by Bacillus (83% of the community) followed by Yersinia (11%) and Escherichia (6%), whereas Mycobacterium was almost undetectable in the community. Perhaps, this can be explained by differences in the copy number of 16S rRNA molecule per cell among species; for instance, the acid-fast M. smegmatis have only 2 copies of 16S rRNA molecule whereas B. amyloliquefaciens has 10 copies. The gram-negative strains E. coli and Y. enterocolitica have similar 16S rRNA copy number of 7 (Ribosomal Database Project [http://rdp.cme.msu.edu]). Moreover, the underestimation of Mycobacterium perhaps is due to low lysability of this bacterium when only exposed to chemical and mechanical cell disruption methods. In addition, among the 4 spiked species, Mycobacterium has the highest GC content (65.6%)24, which further contributes to low amplification efficiency of this bacterium. In contrast, Bacillus had the lowest GC content (42%)25 followed by Yersinia (47%)26, and Escherichia (50%)27. Our results were showing the same trend (Fig. 3a) and supported that species with low GC content are more amplifiable and thus are promoted during the PCR amplification. We should also take into account that although spiked cells were harvested from their late log cultures they may still contain dead cells that could be amplified using DNA-based method. This should be the major reason for the observed differences between the DNA- and RNA-based methods, as RNA-based method only amplified the live and metabolically active proportion of mixed bacterial cells (Fig. 3a, Supplementary Table S1).
The pattern observed during PMA-based amplification of the same community (all live spiked) was very different from both DNA- and RNA-based methods (Fig. 3a and Supplementary Table S1). As indicated above the spiked water with live bacteria may have contained a number of dead cells or cells with damaged/compromised membranes due to osmotic stress when spiked into HPLC water10, 28 or heating and light exposure during PMA treatment29. This could have facilitated the penetration of PMA into such cells, and hence, excluding them as live cells. Hellein et al.30 in spiked environmental waters tested with PMA by qPCR reported that PMA can penetrate into membrane-compromised bacteria even though they are live. The abovementioned stressors perhaps may sensitize bacterial cells especially those of gram-negative bacteria resulting in suppression of these species in the sequenced mini-microbial community of all live spiked cells.
The above-mentioned mechanisms also explain the observed patterns of microbiota when a combination of live and dead cells was spiked. Regardless of Gram status of spiked cells, RNA-based method was superior to DNA- and PMA-based methods in distinguishing live cells although the proportion of these cells were over- or underestimated (Fig. 1, Fig. 3c,d, Supplementary Table S1). For spiked water with live gram-negative and dead gram-positive/acid-fast bacteria, the RNA-based method did not detect the dead gram-positive bacteria, while DNA- and PMA-based methods did (15% in DNA-based and 2% in PMA-based; Fig. 3c). However, the proportion of Yersinia was overestimated (91%) and Escherichia was underestimated (9%) in the community. This might be in one hand due to the higher GC content of Escherichia and reduced amplification efficiency for this species31, but more importantly due to misclassification of OTUs associated with these two species due to the similarity of 16S rRNA in members of Enterobacteriaceae family32. Similarly, for spiked water with live gram-positive/acid-fast and dead gram-negative bacteria, the RNA-based method did not detect the dead gram-negative species but overestimated Bacillus proportion (96%) and underestimated Mycobacterium (4%) most likely due to lower GC content of Bacillus. Compared to RNA-, DNA- and PMA-based methods estimated that 5% and 1% of the community to be associated with dead species, respectively (Figs 1 and 3d). Other studies12, 23, 29, 33 have similarly reported that the DNA from dead/membrane-compromised gram-negative and/or gram-positive cells were not completely removed by PMA treatment, and therefore detected by qPCR analysis.
For the five environmentally relevant water sources, Illumina paired-end sequencing generated on average 27,727 of high quality sequences per sample. An even depth of 11,000 sequences per sample was used for comparison of α-diversity measures among water sources and DNA-, PMA- and RNA-based methods (Fig. 5). Regardless of water source, the Chao1 richness of bacterial community was highest for the DNA-based approach and lowest for RNA-based approach (P < 0.05), with the exception of red river water. The α-diversity metrics differ in the way they account for rare and abundant species. In case of Chao1, it estimates the number of undiscovered species, and as DNA-based method does not differentiate between live and dead bacteria, it identifies more OTUs and thus overestimates the community richness.

α-diversity indices of microbiota of water samples collected from different sources and assessed by DNA-, PMA, and RNA-based 16S rRNA MiSeq Illumina sequencing.
The β-diversity among water sources and DNA-, PMA- and RNA-based methods were visualized and compared using PCoA and PERMANOVA (Fig. 6, Supplementary Table S2). The RNA-based method separated from the DNA- and PMA-based methods particularly in lake and river water that contained proportionally larger amounts of live and dead bacterial cells than the treated water.

Principal coordinates analysis (PCoA) of unweighted UniFrac distances of water samples collected from different sources to compare microbiota β-diversity between DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing.
The average numbers of sequences in five environmentally relevant water sources are presented in Fig. 7. The taxonomic classification of clustered OTUs for these samples revealed the presence of 47 bacterial phyla. Phyla with abundances greater than 0.1% of the community were used to compare the microbiota composition generated using DNA-, PMA-, and RNA-based methods (Fig. 8). The DNA- and PMA-based methods showed a close agreement in phyla, whereas the RNA-based method showed pronounced differences from the DNA- and PMA-based methods for the lake and river samples, and the treated water collected in the water treatment plant (Fig. 8).

Average number of sequences in water samples collected from different sources analyzed using DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing. *P < 0.05.

Microbiota compositions in water samples collected from different sources determined using DNA-, PMA-, and RNA-based 16S rRNA MiSeq Illumina sequencing.
As indicated earlier optimization of PMA-based sequencing for capturing live members of microbial community in water samples has major difficulties. Different proportions of gram-negative, gram-positive and acid-fast bacteria are present within each community at different physiological growth stages and PMA reaction with DNA from these communities differ. Several studies12, 23, 29, 33 reported that PMA-based qPCR may be less effective for samples with a complex mixture of gram-positive and gram-negative bacterial cell, or when the ratio of dead to live cells was high.
Our study supports these observations and recommends RNA-based approach as a superior tool for proper estimation of live (either dormant or metabolically active) members of bacterial community in the water. That being said, it should be noted that RNA is less stable than DNA and might be more difficult to purify from certain environmental samples34,35,36. Our data also recognizes that the abundances of the community members are always overestimated or underestimated due to biases introduced during the DNA or RNA extraction steps (e.g. due to lisability of cells and Gram status) and differential amplification of target during PCR (e.g. due to GC content and primer efficiency). These biases will continue to be a challenge for microbial community analyses regardless of DNA-based or RNA-based 16S rRNA sequencing method of choice.
Materials and Methods
Bacterial strains and culture condition used for spiking water
Gram-positive Bacillus amyloliquefaciens (strain BS6), acid-fast Mycobacterium smegmatis and two gram-negative strains, Escherichia coli and Yersinia enterocolitica, obtained from the Microbiome Laboratory (Department of Animal Science, University of Manitoba, Canada) were cultured in the liquid Luria-Bertani media (LB; Difco, Fisher Scientific, Edmonton, AB, Canada) on a rotary shaker (New Brunswick Scientific, Edison, NJ, USA). E. coli and Y. enterocolitica were cultured at 200 rpm and 37 °C for 16–18 h. M. smegmatis was cultured at 200 rpm and 37 °C for 72 h. B. amyloliquefaciens was cultured at 180 rpm and 30 °C for 16 to 18 h. All bacteria were collected from late log cultures to minimize the number of dead bacteria within the cultures.
For each of the cultured species, half of the culture was killed by placing flasks in a water bath at 95 °C for 30 min and confirming that aliquots (100 μl) did not produce any visible colonies on the LB agar (Difco) upon incubation. To experimentally create mini-microbial communities, ultra-pure HPLC-grade (Fisher Scientific, Ottawa, ON, Canada) was used as the water source and spiked with mixtures of bacterial species present at a concentration of 1.5 × 104 CFU/ml in triplicate as follow: (1) four live bacterial species, (2) four dead bacterial species, (3) dead gram-positive and acid-fast species plus live gram-negative species, (4) dead gram-negative species plus live gram-positive and acid-fast species, and (5) HPLC grade water without any bacterial culture (negative control).
Environmental water sample collection
Water samples (n = 3 per treatment group) were collected from a First Nation community in Northern Manitoba in July 2014, and in the City of Winnipeg in September 2014 using standard methods37: SM9060A (sample bottle pretreatment) and SM 9060B (preservation and storage). Water samples in the Northern community included: 1) tap water from homes that are served by piped water from the water treatment plant (0–3 CFU/100 ml total coliforms, and 0–2 CFU/100 ml E. coli), 2) tap water from homes which receive potable water delivered from the water treatment plant by a water truck to a cistern (water holding tank) (2–430 CFU/100 ml total coliforms, and 1–400 CFU/100 ml E. coli), and 3) samples collected from the lake that is the source water to the water treatment plant (50–690 CFU/100 ml total coliforms, and 0–330 CFU/100 ml E. coli). Samples were transported in coolers to Winnipeg by air on the same day of collection, or in the next morning after stored overnight in a refrigerator. Water samples in the City of Winnipeg included 1) tap water from homes that are served by piped water from the City’s water treatment plant (0 CFU/100 ml total coliforms or E. coli), and 2) samples collected from the Red River flowing through the city (27,000–37,000 CFU/100 ml total coliforms, and 0–1,000 CFU/100 ml E. coli).
All collected samples were immediately processed upon receiving in the level 2 biological safety Microbiome Laboratory (Department of Animal Science, University of Manitoba, Canada). This processing included determinations of total coliform and E. coli counts using the standard membrane filter procedure as outlined in SM922237. Briefly, 100 ml of water sample was filtered through a sterile 0.20 µm Polyethersulfone membrane filter (Mo Bio Laboratories, Inc. West Carlsbad, CA, USA). The filter paper was placed on agar plates containing chromogenic Brilliant E. coli and coliform medium (Fisher Scientific, Ottawa, ON, Canada) and incubated at 35 °C for 24 h. Some samples required dilution as bacterial counts were too high.
Extraction of DNA and RNA from water samples and DNA from PMA treated water
For the HPLC graded water spiked with mixtures of bacterial species, aliquots of 100 ml spiked water were filtered through 0.20 μm Polyethersulfone filter (Mo Bio Laboratories Inc., West Calsbad, CA, USA). For the environmental water samples, 500 ml aliquots were filtered through 0.20 μm Polyethersulfone filter (Mo Bio). DNA and RNA were extracted using Powerwater DNA and RNA isolation kits (Mo Bio), respectively, that included a bead-beating step for mechanical lysis of bacteria. DNase I treatment step was included during RNA extraction for removal of DNA. RNA was transcripted to cDNA using a reverse transcription kit (QuantiTec, Qiagen Scientific, Germantown, MD, USA). For the PMA treatment, a modified method based on Hellein et al.30 and Nocker et al.10 was used. In brief, following filtering of 500 ml aliquots of water, 5 μl PMA was added onto the filter when 2 ml water was left, to give a final PMA concentration of 50 μm (PMA dye, Biotium, Inc., Hayward, CA, USA). The PMA spread evenly across the filter. The filters were incubated on a rocker with aluminum foil for 20 min and then placed on an ice block set 20 cm from a light source. They were exposed for 15 min to 500 W Halogen light to cross-link the PMA to DNA. DNA was extracted immediately after PMA treatment using MoBio Powerwater DNA kit. Triplicate samples were processed for each defined mixture of spiked water. The quantity of the DNA and cDNA were measured using NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). The quality was measured by PCR amplification of 16S rRNA, using forward primer 27 F (AGAGTTTGATCMTGGCTCAG) and reverse primer 342 R (CTGCTGCSYCCCGTAG), after DNA and cDNA were normalized to the concentration of 10 ng/μl38.
Library construction and Illumina sequencing
A library of V4 region of 16S rRNA was constructed using modified F515 and R806 primers39 as described previously40. PCR reaction for each sample was performed in duplicate and contained 1.0 µl of pre-normalized DNA, 1.0 µl of each forward and reverse primers (10 µM), 12 µl HPLC grade water (Fisher Scientific, Ottawa, ON, Canada) and 10 µl 5 Prime Hot MasterMix (5 Prime, Inc., Gaithersburg, MD, USA). Reactions consisted of an initial denaturing step at 94 °C for 3 min, followed by 35 amplification cycles at 94 °C for 45 sec, 50 °C for 60 sec, and 72 °C for 90 sec; finalized by an extension step at 72 °C for 10 min in an Eppendorf Mastercycler pro (Eppendorf, Hamburg, Germany). Next, PCR products were purified using ZR-96 DNA Clean-up Kit (ZYMO Research, Irvine, CA, USA) to remove primers, dNTPs and reaction components. The V4 library was then generated by pooling 200 ng of each sample, quantified by Picogreen dsDNA (Invitrogen, Burlington, NY, USA). The pooled library was diluted to a final concentration of 5 pM using pre-chilled hybridization buffer (HT1) (Illumina, Irvine, CA, USA) measured by Qubit 2.0 Fluorometer (Life technologies, Ottawa, ON, Canada). In the final step, 15% of PhiX control library was spiked into the amplicon library to improve the unbalanced and biased base composition. For Illumina sequencing, customized sequencing primers for read1 (5′-TATGGTAATTGTGTGCCAGCMGCCGCGGTAA-3′), read2 (5′-AGTCAGTCAGCCGGACTACHVGGGTWTCTAAT-3′) and index read (5′-ATTAGAWACCCBDGTAGTCCGGCTGACTGACT-3′), and mixture of sample and PhiX library were added to the MiSeq Reagent Kit V2 (300-cycle; Illumina, San Diego, CA, USA). The 150 paired-end sequencing reaction was performed on a MiSeq Illumina platform at the Microbiome Laboratory (Department of Animal Science, University of Manitoba, Canada). The sequence data are uploaded into the Sequence Read Archive (SRA) or NCBI (http://www.ncbi.nlm.nih.gov/sra) and accessible through accession number SRR2983316.
Bioinformatics and statistical analysis
The FLASH assembler41 was used to merge overlapping paired-end Illumina fastq files. All the sequences with mismatches or ambiguous calls in the overlapping region were discarded. The output of fastq file was then analyzed by downstream computational QIIME pipelines42. Assembled reads were de-multiplexed according to barcode sequences, chimeric reads were filtered using UCHIME43 and sequences were assigned to Operational Taxonomic Units (OTU) using the QIIME implementation of UCLUST44 at 97% pairwise identity threshold. Taxonomies were classified to the representative sequence of each OTU using RDP classifier45 and aligned with the Greengenes Core reference database46 using PyNAST algorithms47. A phylogenetic tree was built with FastTree 2.1.3.48 for further comparisons between microbial communities.
Within-community diversity (α-diversity) was calculated by different indices of species richness and evenness including Shannon, Simpson, Chao1, and observed number of species using the open source software QIIME. Permutational analysis of variance (PERMANOVA, Anderson, 2005) and principal coordinates analysis (PCoA) of unweighted UniFrac distance matrices49 were used to assess between-sample differences in microbiota diversity and community structures (Warwick and Clarke, 2006). The differences of α-diversity indices and number of sequences among treatments were tested using MIXED procedure (Tukey studentized range adjustment) of SAS (version 9.3, SAS Institute Inc., Cary, NC, USA) with the effect of sequencing method as the fixed factor. For non-normally distributed data, GLIMMIX procedure of SAS fitted with Poisson or negative binomial distributions was used. The differences between treatments were considered significant at P < 0.05.
Change history
22 November 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Ramirez-Castillo, F. Y. et al. Waterborne pathogens: detection methods and challenges. Pathogens 4, 307–334, doi:10.3390/pathogens4020307 (2015).
Feazel, L. M. et al. Opportunistic pathogens enriched in showerhead biofilms. Proc Natl Acad Sci USA 106, 16393–16399, doi:10.1073/pnas.0908446106 (2009).
Proctor, C. R. & Hammes, F. Drinking water microbiology–from measurement to management. Curr Opin Biotechnol 33, 87–94, doi:10.1016/j.copbio.2014.12.014 (2015).
Barcina, I. & Arana, I. The viable but nonculturable phenotype: a crossroads in the life-cycle of non-differentiating bacteria? Reviews in Environmental Science and Bio/Technology 8, 245–255 (2009).
Lagier, J. C. et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 1, 16203, doi:10.1038/nmicrobiol.2016.203 (2016).
Nielsen, K. M., Johnsen, P. J., Bensasson, D. & Daffonchio, D. Release and persistence of extracellular DNA in the environment. Environ Biosafety Res 6, 37–53, doi:10.1051/ebr:2007031 (2007).
Haack, S. K. et al. Genes Indicative of Zoonotic and Swine Pathogens Are Persistent in Stream Water and Sediment following a Swine Manure Spill. Appl Environ Microbiol 81, 3430–3441, doi:10.1128/AEM.04195-14 (2015).
Selenska, S. & Klingmuller, W. DNA recovery and direct detection of Tn5 sequences from soil. Lett Appl Microbiol 13, 21–24 (1991).
Nocker, A., Cheung, C. Y. & Camper, A. K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods 67, 310–320, doi:10.1016/j.mimet.2006.04.015 (2006).
Nocker, A., Richter-Heitmann, T., Montijn, R., Schuren, F. & Kort, R. Discrimination between live and dead cellsin bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol 13, 59–65, doi:10.2436/20.1501.01.111 (2010).
Vaishampayan, P. et al. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J 7, 312–324, doi:10.1038/ismej.2012.114 (2013).
Varma, M. et al. Quantitative real-time PCR analysis of total and propidium monoazide-resistant fecal indicator bacteria in wastewater. Water Res 43, 4790–4801, doi:10.1016/j.watres.2009.05.031 (2009).
Gensberger, E. T. et al. Evaluation of quantitative PCR combined with PMA treatment for molecular assessment of microbial water quality. Water Res 67, 367–376, doi:10.1016/j.watres.2014.09.022 (2014).
Rogers, G. B. & Bruce, K. D. Next-generation sequencing in the analysis of human microbiota: essential considerations for clinical application. Mol Diagn Ther 14, 343–350, doi:10.2165/11587680-000000000-00000 (2010).
Fittipaldi, M., Nocker, A. & Codony, F. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J Microbiol Methods 91, 276–289, doi:10.1016/j.mimet.2012.08.007 (2012).
Bleve, G., Rizzotti, L., Dellaglio, F. & Torriani, S. Development of reverse transcription (RT)-PCR and real-time RT-PCR assays for rapid detection and quantification of viable yeasts and molds contaminating yogurts and pasteurized food products. Appl Environ Microbiol 69, 4116–4122 (2003).
Morin, N. J., Gong, Z. & Li, X. F. Reverse transcription-multiplex PCR assay for simultaneous detection of Escherichia coli O157:H7, Vibrio cholerae O1, and Salmonella Typhi. Clin Chem 50, 2037–2044, doi:10.1373/clinchem.2004.036814 (2004).
Hambraeus, G., von Wachenfeldt, C. & Hederstedt, L. Genome-wide survey of mRNA half-lives in Bacillus subtilis identifies extremely stable mRNAs. Mol Genet Genomics 269, 706–714, doi:10.1007/s00438-003-0883-6 (2003).
Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M. & Rosenow, C. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res 13, 216–223, doi:10.1101/gr.912603 (2003).
Cornell, J. A. Experiments with Mixtures: Designs, Models, and the Analysis of Mixture Data. Third Edition edn, (John Wiley & Sons, 2011).
Brooks, J. P. et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15, 66, doi:10.1186/s12866-015-0351-6 (2015).
Pacheco, S. A., Hsu, F. F., Powers, K. M. & Purdy, G. E. MmpL11 protein transports mycolic acid-containing lipids to the mycobacterial cell wall and contributes to biofilm formation in Mycobacterium smegmatis. J Biol Chem 288, 24213–24222, doi:10.1074/jbc.M113.473371 (2013).
Elizaquivel, P., Sanchez, G. & Aznar, R. Quantitative detection of viable foodborne E. coli O157:H7, Listeria monocytogenes and Salmonella in fresh-cut vegetables combining propidium monoazide and real-time PCR. Food Control 25, 704–708 (2012).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544, doi:10.1038/31159 (1998).
Muto, A. & Osawa, S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci USA 84, 166–169 (1987).
Moore, R. L. & Brubaker, R. R. Hybridization of Deoxyribonucleotide Sequences of Yersinia-Enterocolitica and Other Selected Members of Enterobacteriaceae. Int J Syst Bacteriol 25, 336–339 (1975).
Blattner, F. R. et al. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 (1997).
Bogosian, G. et al. Death of the Escherichia coli K-12 strain W3110 in soil and water. Appl Environ Microbiol 62, 4114–4120 (1996).
Lovdal, T., Hovda, M. B., Bjorkblom, B. & Moller, S. G. Propidium monoazide combined with real-time quantitative PCR underestimates heat-killed Listeria innocua. J Microbiol Methods 85, 164–169, doi:10.1016/j.mimet.2011.01.027 (2011).
Hellein, K. N. et al. A filter-based propidium monoazide technique to distinguish live from membrane-compromised microorganisms using quantitative PCR. J Microbiol Methods 89, 76–78, doi:10.1016/j.mimet.2012.01.015 (2012).
McDowell, D. G., Burns, N. A. & Parkes, H. C. Localised sequence regions possessing high melting temperatures prevent the amplification of a DNA mimic in competitive PCR. Nucleic Acids Res 26, 3340–3347 (1998).
Sanderson, K. E. Genetic relatedness in the family Enterobacteriaceae. Annu Rev Microbiol 30, 327–349, doi:10.1146/annurev.mi.30.100176.001551 (1976).
Pan, Y. & Breidt, F. Enumeration of viable Listeria monocytogenes cells by real-time PCR with propidium monoazide and ethidium monoazide in the presence of dead cells. Appl Environ Microb 73, 8028–8031, doi:10.1128/Aem.01198-07 (2007).
Hierro, N., Esteve-Zarzoso, B., Gonzalez, A., Mas, A. & Guillamon, J. M. Real-time quantitative PCR (QPCR) and reverse transcription-QPCR for detection and enumeration of total yeasts in wine. Appl Environ Microb 72, 7148–7155, doi:10.1128/Aem.00388-06 (2006).
Andorra, I., Esteve-Zarzoso, B., Guillamon, J. M. & Mas, A. Determination of viable wine yeast using DNA binding dyes and quantitative PCR. Int J Food Microbiol 144, 257–262, doi:10.1016/j.ijfoodmicro.2010.10.003 (2010).
Blazewicz, S. J., Barnard, R. L., Daly, R. A. & Firestone, M. K. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. Isme Journal 7, 2061–2068, doi:10.1038/ismej.2013.102 (2013).
Rice, E. W., Baird, R. B., Eaton, A. D. & Clesceri, L. S. Standard Methods for the Examination of Water and Wastewaters. 22nd ed edn, (American Public Health Association, American Water Works Association, Water Environment Federation, 2012).
Khafipour, E., Li, S., Plaizier, J. C. & Krause, D. O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl Environ Microbiol 75, 7115–7124, doi:10.1128/AEM.00739-09 (2009).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624, doi:10.1038/ismej.2012.8 (2012).
Derakhshani, H., Tun, H. M. & Khafipour, E. An extended single-index multiplexed 16S rRNA sequencing for microbial community analysis on MiSeq illumina platforms. J Basic Microbiol 56, 321–326, doi:10.1002/jobm.201500420 (2016).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology 73, 5261–5267 (2007).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology 72, 5069–5072 (2006).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one 5, e9490 (2010).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology 71, 8228–8235 (2005).
Acknowledgements
The research was funded by CREATE H2O grant from the Natural Science and Engineering Council of Canada and University of Manitoba Start-up funds.
Author information
Authors and Affiliations
Contributions
A.F., E.K., A.K., and W.G.D.F. conceived and designed the experiment. R.L. and H.M.T. conducted the experiment and performed lab analyses. R.L., H.M.T., M.J., Z.Z. and E.K. developed the bioinformatics and statistical models and analyzed the data. All authors drafted the manuscript. All authors carefully read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests..
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, R., Tun, H.M., Jahan, M. et al. Comparison of DNA-, PMA-, and RNA-based 16S rRNA Illumina sequencing for detection of live bacteria in water. Sci Rep 7, 5752 (2017). https://doi.org/10.1038/s41598-017-02516-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02516-3
Keywords
This article is cited by
-
Laser capture microdissection as a method for investigating the human hair follicle microbiome reveals region-specific differences in the bacteriome profile
BMC Research Notes (2023)
-
Aerial transport of bacteria by dust plumes in the Eastern Mediterranean revealed by complementary rRNA/rRNA-gene sequencing
Communications Earth & Environment (2023)
-
Impact of nutrients and trace elements on freshwater microbial communities in Croatia: identifying bacterial bioindicator taxa
Environmental Science and Pollution Research (2023)
-
Protists modulate active bacterial community composition in paddy field soils
Biology and Fertility of Soils (2023)
-
Nasal microbiome disruption and recovery after mupirocin treatment in Staphylococcus aureus carriers and noncarriers
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
