
Abstract
Tyrosinases are an ubiquitous group of copper containing metalloenzymes that hydroxylate and oxidize phenolic molecules. In an application context the term ‘tyrosinase’ usually refers to ‘mushroom tyrosinase’ consisting of a mixture of isoenzymes and containing a number of enzymatic side-activities. We describe a protocol for the efficient heterologous production of tyrosinase 4 from Agaricus bisporus in Escherichia coli. Applying this procedure a pure preparation of a single isoform of latent tyrosinase can be achieved at a yield of 140 mg per liter of autoinducing culture medium. This recombinant protein possesses the same fold as the enzyme purified from the natural source as evidenced by single crystal X-ray diffraction. The latent enzyme can be activated by limited proteolysis with proteinase K which cleaves the polypeptide chain after K382, only one The latent enzyme can amino acid before the main in-vivo activation site. Latent tyrosinase can be used as obtained and enzymatic activity may be induced in the reaction mixture by the addition of an ionic detergent (e.g. 2 mM SDS). The proteolytically activated mushroom tyrosinase shows >50% of its maximal activity in the range of pH 5 to 10 and accepts a wide range of substrates including mono- and diphenols, flavonols and chalcones.
Similar content being viewed by others

Introduction
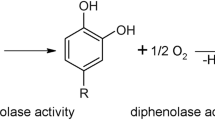
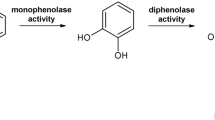
Tyrosinases form an ubiquitous family of metalloenzymes and are found in all domains of life1. They are of central importance for the pigmentation in vertebrates as the reactions catalyzed by tyrosinase provide the starting material for melanin biosynthesis2. Tyrosinases catalyze the ortho-hydroxylation of monophenols to o-diphenols (monophenolase or cresolase activity, EC 1.14.18.1) as well as the subsequent two-electron oxidation to the respective o-quinones (diphenolase or catechol oxidase activity, EC 1.10.3.1), which is coupled with the reduction of molecular oxygen to water3, 4. The active site of tyrosinase is composed of two copper ions which are coordinated by three histidine side chains each5 forming a type III copper center6. Activation of molecular oxygen is affected by binding of dioxygen to the type III copper center in a characteristic ‘side-on’ bridging mode (µ-η2:η2)7,8,9. Hydroxylation and oxidation of one monophenol to the corresponding o-quinone requires one molecule of O2, while two o-diphenols can be oxidized to o-quinones per molecule of O2 consumed (see Fig. 1).

Tyrosinase catalyzes the o-hydroxylation of monophenols (top) and the oxidation of o-diphenols (bottom).
The products of the reactions catalyzed by tyrosinase – o-quinones – are highly reactive and consequently commonly unstable in the biological environment they are generated in10. Therefore, they do participate in a number of non-enzymatic, spontaneous reactions. The best studied among those reactions are the formation of high-molecular-weight adducts, especially melanin10,11,12,13,14, Michael-type nucleophilic 1,4-additions15, 16 and the direct coupling of two quinones (‘phenol coupling’)15, 17, 18. As tyrosinases can oxidize both small phenolic molecules and phenolic moieties of larger molecules (e.g. proteins) they have found a plethora of biotechnological applications in e.g. organic synthesis, determination of phenolic analytes, bioremediation as well as in medicine, food processing and engineering of (bio)materials19, 20.
Most of these application utilize tyrosinase isolated from fruiting bodies of the common white mushroom Agaricus bisporus (‘mushroom tyrosinase’)20, supposedly mainly due to its ready commercial availability21. However, it should be noted that the purification protocols used to prepare the available commercial preparations do usually not yield homogenous tyrosinase and these are therefore likely to contain one or more unspecified ‘extras’ like laccase, β-glucosidase, β-xylosidase, cellulase, chitinase and xylanase activities22, 23. The purity of these preparations is further compromised by the fact that Agaricus bisporus possesses genes coding for six different tyrosinases (AbPPO1 - AbPPO6)24, 25. Of those six genes, at least two (AbPPO3 and AbPPO4)25,26,27 are expressed in significant amounts in the fruiting bodies which serve as the source material for the commercial preparations of ‘mushroom tyrosinase’. Considering these limitations of the widely used tyrosinase preparations in terms of both purity and batch-to-batch variability, an alternative enzyme source that manages to provide a single isoform of tyrosinase in a pure form and at a constant quality would promote both basic (protein-)biochemical research and biotechnological applications of tyrosinase. Herein, we describe such a protocol, yielding pure AbPPO4 in its latent form as well as providing access to the mature, active form of the enzyme.
Results
Sequence of AbPPO4
The initial PCR on the A. bisporus cDNA with primers enframing the ORF for AbPPO4 (AbPPO4_fwd and AbPPO4_rev) yielded two distinct bands at 1.8 kbp (expected size of the gene) and 1.4 kbp (Figure S3). By cloning and sequencing both molecules were identified as the expected gene for AbPPO4. While the larger one (1836 bp) contained the complete gene encoding amino acids M1 to F611, the smaller band (1401 bp) was missing 435 bp corresponding to 145 amino acids. The loss of these bases occurred in such a manner that the reading frame was conserved and amino acids A436 to A580 were deleted from the translation product. This deletion starts in the middle of exon number 6 and spans the last intron as well as more than two-thirds of the last exon in the gene for AbPPO4. On the protein level the missing amino acids are all located in the C-terminal domain and contain the CXXC-motif as well as approximately half of the putative trans-membrane helix27. Expression of this construct was attempted but for all the conditions tested the heterologous protein was found exclusively in the insoluble fraction and no tyrosinase activity could be detected in the cell lysate.
With respect to the published sequence for the AbPPO4-gene28 the cloned full-length gene contains 23 mutations among which four are non-silent (see Table 1). Of those two are located in the main domain and two at the end of the C-terminal domain. The two variations in the main domain are already known to be compatible with enzymatic activity as they were also encountered in the enzyme purified from the natural source27. Those and the two remaining changed amino acids as well as all the changes to the amino acid sequence in the second sequence (with the exception of the deletion of 145 amino acids) are predicted to be non-detrimental to the enzyme’s function by SIFT29. Expression of the full-length construct (up to F611) confirms this prediction as the heterologous enzyme was found to be fully active.
Expression and purification of AbPPO4
The latent form of the tyrosinase (up to T565)27 was expressed as a fusion protein with an N-terminal tag, namely glutathione-S-transferase from Schistosoma japonicum 30. Initial expression attempts employing induction by addition of IPTG yielded a big amount of heterologous protein which was however almost exclusively in an insoluble form. Cultivation at lower temperatures and the use of autoinduction medium31 did produce a very small fraction of fusion protein in soluble form so that enzymatic activity could be detected in the cell lysate after 3 days of reaction. The addition of 500 mM NaCl to the cultivation medium did decrease the specific growth rate of the production strain by 30% causing a marked increase in total cultivation time but did also increase the fraction of soluble protein by several orders of magnitude. This expression protocol as described in Methods yields around 200 mg of fusion protein per liter of medium after capture and purification by affinity chromatography employing the affinity of GST to glutathione immobilized on cross-linked agarose. Removal of the fusion partner by the specific protease HRV 3 C was usually quite efficient with yields between 80% and 100% corresponding to approximately 110 to 140 mg of latent tyrosinase per liter of expression culture. The preparations were still active after 1 year of storage at 4 °C in the used 10 mM HEPES pH 7.5 buffer. For long-term storage freezing in liquid nitrogen32 is recommended as the preparations tend to darken after a few months of storage. This darkening, however, does not cause the loss of enzymatic activity.
While the full-length construct did behave almost identical as the latent version of AbPPO4 in heterologous expression, the construct encoding only the main domain of the tyrosinase (up to S383)27 did not exhibit any tyrosinase activity.
Activation of latent AbPPO4
Proteolytic activation of the latent tyrosinase was tested with trypsin and proteinase K and both proteases induced tyrosinase activity in the latent enzyme. The activation by proteinase K was more efficient and did yield an essentially pure preparation of active tyrosinase while even after prolonged incubation with trypsin a second species of approximately 52 kDa remained in the reaction mixture (Figure S4). The digestion with an unspecific protease resulted in a low yield of the activation of 25–50% corresponding to 19–34 mg per liter of culture media. The activated protein appeared as a single peak in the size exclusion chromatogram and had a purity of >95% as estimated from SDS-PAGE (Fig. 2).

SDS-PAGE (10–15% acrylamide) of the four purification stages. L: cell lysate, F: flow-through of the first affinity chromatography step, P: insoluble cell material, M: molecular weight marker; Weights of the used standard protein are indicated left of the gel., AC: eluate of the first affinity chromatography step, l: latent AbPPO4 (eluate of the second affinity chromatography step), a: activated AbPPO4 (eluate of size exclusion chromatography after activation with proteinase K).
Enzymatic activity could be induced in the latent tyrosinase by treatment with ionic detergents. The cationic detergent cetylpyridinium chloride (CPC) was more effective in activating latent AbPPO4 than the anionic sodium dodecyl sulfate (SDS). Maximal activation was achieved by applying 0.1 mM of CPC and reached a level which was 25% higher than that at the optimal SDS-concentration of 0.6 mM (see Fig. 3). The activity level for activation by CPC remained fairly constant at concentrations higher than 0.1 mM but above 2 mM the latent tyrosinase was prone to precipitation. For SDS the enzymatic activity decreased slightly above 0.6 mM but remained constant at circa 90% of the maximal level for up to 5 mM SDS. Above this concentration the enzymatic activity did slowly decrease but no protein precipitation was observed even at SDS levels above 100 mM.

Activation of latent AbPPO4 by ionic detergents. One data point represents the average of three activity measurements on 1 mM L-tyrosine with the indicated concentration of detergent in the respective assay mixture. The error bars indicate ± one standard deviation.
Analysis of the purified tyrosinase by intact protein mass spectrometry
ESI-MS of the acidified (causes the loss of copper)27 but otherwise intact protein (see Fig. 4) yielded a mass of 65096.7 ± 0.30 Da for the latent tyrosinase which matches the calculated mass of 65096.46 Da for the complete sequence (see Fig. 5) from the N-terminal glycine at position −8 to the C-terminal threonine 565 with one thioether bridge (−2.016 Da) and one closed disulfide bridge (−2.016 Da). The presence of a closed disulfide bridge was also reported for the enzyme isolated from the natural source after electrospray ionisation in positive mode27 but the disulfide bridge was found in the open form in the crystal structure33. The only free cysteine residues in the protein are very close to each other in the C-terminal domain of the enzyme (C462 and C465). Since positive electrospray ionisation of free cysteine has been shown to generate mainly protonated cystine (which was attributed to oxidation processes occurring during positive mode electrospray)34 a similar mechanism may also be observed for the pseudo molecular ions of AbPPO4. The mass spectra of the activated tyrosinase indicated the presence of two protein variants with determined masses of 44181.5 ± 0.51 Da and 44449.6 ± 0.48 Da, respectively. These masses indicate proteolytic cleavage after lysine 382 (calculated: 44449.35 Da) and additionally for most of the tyrosinase molecules the removal of the first three N-terminal amino acids (Gly-Pro-Leu) from the vector-derived region (calculated: 44182.03 Da).

ESI-MS of latent and activated AbPPO4; (A) latent AbPPO4, (B) activated AbPPO4.

Sequence of the expressed AbPPO4. shaded in grey: vector-derived sequence; blue: central domain up to serine 383, containing the copper-coordinating histidines (red); yellow: cysteine 80 which forms a thioetherbridge with histidine 82; orange: C-terminal domain up to T565 (C-terminus of the main expressed construct); green: CXXC-motif containing all the free cysteine residues of the enzyme; The four amino acid mutations relative to C7FF05 (Table 1) are underlined and marked in bold.
Crystal structure of latent AbPPO4
Latent tyrosinase crystallized in the monoclinic space group C 1 2 1 with 4 chains in the asymmetric unit giving rise to a unit cell of a = 287.30 Å, b = 52.09 Å, c = 152.66 Å, α = 90.00°, β = 98.03° and γ = 90.00°. The obtained crystals diffracted to a resolution of 3.25 Å (for further statistics see Table S2). In contrast to the crystals obtained with the enzyme isolated from the natural source which contained two different chains in the asymmetric unit (one latent and one active protein)33 and did only form in the presence of sodium hexatungstotellurate(VI) (Na6[TeW6O24] • 22 H2O, TEW)35, 36, the recombinant enzyme formed crystals containing exclusively the latent tyrosinase. Inspection of the electron density gave no indication for any deviation from the sequence shown in Fig. 5. The recombinant enzyme assumes the same fold as the tyrosinase isolated from the natural source and their active centers as well as the surrounding amino acids are virtually identical (see Fig. 6).

Alignment of AbPPO4 isolated from the natural source and the recombinant enzyme. The latent chain of AbPPO4 isolated from Agaricus bisporus (PDB 4OUA, chain B; shown in blue)36 was aligned with the heterologously produced protein (PDB 5M6B, chain B; shown in red). The r.m.s.d. between the positions of 3616 matched atoms amounts to 0.435 Å.
The amino acid side chains which form hydrogen bonds with the TEW polyoxyanion in the crystal of the enzyme purified from the natural source (HKKE starting at H116) are found in equivalent positions in the crystal of the recombinant enzyme but the lysine side chains are considerably more flexible as indicated by the lack of electron density beyond the Cβ-atoms. In the crystal containing TEW the interactions mediated by TEW are crucial for the stacking of the monomer chains while in the crystal of recombinant latent AbPPO4 this motif does not play a significant role for the formation of the lattice with the closest interchain contact being longer than 6 Å. The missing contribution from these strong interactions may be one decisive factor limiting the attainable crystallographic resolution35, 37 which is 0.49 Å worse than in the crystal structure obtained using TEW.
Characterization of the activated AbPPO4
The optimal pH for the enzymatic conversion of L-tyrosine was found to be 6.8 (Figure S5). Enzymatic activity is found starting from pH 4 and extends beyond pH 10.5. In the pH-range from 5 to 10 the enzyme retains more than 50% of its activity at the optimal pH. The activated AbPPO4 catalyzes both reactions observed for tyrosinase (Fig. 1). The catechol oxidase activity proceeds typically with a rate two orders of magnitude faster than the hydroxylation and oxidation of monophenols (Table 2 and Figure S7). Of the tested substrates the enzyme exhibits the highest affinity and the lowest reaction rate for L-tyrosine. The reaction with catechol shows a decrease in rate for catechol concentrations higher than 10 mM which is probably due to the effective suicide-inactivation of tyrosinase by this substrate38, 39. Evidence for such a loss of activity during catalysis has also been presented for the type III copper enzyme aurone synthase from Coreopsis grandiflora Coreopsis grandiflora acting on sulfuretin (which does contain a catechol moiety)40.
Besides the substrates chosen for kinetic measurements AbPPO4 was also tested with and does accept tyrosol, chlorogenic acid, p-coumaric acid, 4-tert-butlycatechol, octopamine, 4-methylcatechol, resorcinol, hydroquinone, protocatechuic acid, 3,4-dihydroxyphenylacetic acid, pyrogallol and 3-methoxyphenol as well as the flavonols fisetin, quercetin, its glycoside rutin, the flavanone naringenin and the chalcons isoliquiritigenin and butein (see Figure S6 and Figure S8 for structural formulas of substrates accepted by AbPPO4).
Activated AbPPO4 discriminates between enantiomers of tyrosine showing pronounced differences in the rate of the tyrosinase reaction. For tyrosine 1 mM of the L-enantiomer is converted at a rate of 1.22 ± 0.0073 U mg−1, which is 2.58 ± 0.020 times faster than the rate on D-tyrosine. A slight increase in enantioselectivity is seen for the methyl ester of tyrosine for which the respective value is 14.3 ± 0.11 U mg−1 for L-tyrosine methyl ester representing a ratio of 3.70 ± 0.050 relative to the rate for the D-enantiomer.
Discussion
For bacterial tyrosinases productivities in the gram per liter range have been demonstrated with the application of an optimized fed-batch strategy41 but for eukaryotic tyrosinases a yield of 4 to 6 mg per liter of culture is already considered large-scale42 and the same value was also reported for a related plant enzyme43. Expression strategies targeting fungal tyrosinases did usually rely on fungal hosts for the expression of soluble and active tyrosinase44,45,46. Substantial expression yields per liter of culture were reported for the secreted TYR2 from Trichoderma reesei overexpressed homologously (1 g l−1 in batch fermentation)45 or in Komagataella pastoris (24 mg l−1)46 and for a tyrosinase from Pycnoporus sanguineus produced heterologously in Aspergillus niger (20 mg l−1)44. As fungi do possess the necessary molecular tools to activate latent tyrosinases all the isolated enzymes were in their active form47. Bacterial expression of fungal tyrosinase does provide access to latent tyrosinases but was hampered by insufficient solubility of the expressed proteins which were also prone to enzymatic inactivity28. Enzymatically functional protyrosinase from Pholiota microspora was expressed in Escherichia coli 48 but no value for the yield was reported. Recently, protyrosinase from Polyporus arcularius was produced in the same host with a yield of 54 mg latent tyrosinase per liter of culture medium49. Here, the latent form of AbPPO4 is produced by E. coli at a yield of 110 to 140 mg per liter of culture. The conversion of latent tyrosinase into the enzymatically active form is coupled to the proteolytic removal of the C-terminal domain which shields the active site of the tyrosinase47, 50. The causal agents for this key-step in the maturation of fungal tyrosinases are still elusive and for only four fungal tyrosinases the exact location of this crucial event is known. Neurospora crassa TYR is activated in vivo by cleavage after F40851, active TYR2 of Trichoderma reesei extends up to G40045, TYR1 from Pholiota nameko is cleaved after F38752 and the C-terminal residue of active AbPPO4 is S38327. All those cleavage sites are found 31 or 30 (T. reesei TYR2) amino acids after the tyrosine motif (Y-X-Y/F or Y/F-X-Y) which is found close to the end of the central domain in all tyrosinases53. Preceding the cleavage site by 4 amino acids, the YG-motif, which is conserved in fungal tyrosinases20, is present. The activated AbPPO4 presented herein was cleaved by proteinase K after K382, which is only one amino acid away from the in vivo activation site S38327. This activated tyrosinase should therefore be an excellent model for the native enzyme. Digestion with proteinase K has also been used as a purification method for tyrosinase from mice54 and apple55. This stability against proteinase K digestion of tyrosinases from three different kingdoms of life suggests resistance against proteolysis by serine proteases as a general feature of tyrosinases.
Besides proteolytic activation enzymatic activity may also be induced in latent tyrosinases by exposing them to acidic conditions56 or detergents like SDS57. Employing such a system, the enzymatic activity may be kept dormant in a preparation for a prolonged period of time until it is needed at which point it may be induced by the simple addition of a detergent. Latent AbPPO4 was activated by both the cationic detergent CPC and the anionic detergent SDS (see Fig. 3). Activation by CPC did yield a circa 25% higher activity than SDS-activation did and required only one sixth of the detergent concentration. For routine analysis during purification we employed SDS (at a concentration of 2 mM) as CPC did cause precipitation of preparations still containing significant concentrations of foreign proteins and, at concentrations above 2 mM, also AbPPO4 itself.
Activated AbPPO4 retains >50% of its activity at the optimal pH 6.8 in the range of pH 5 to pH 10 providing a wide range of possible reaction conditions. For most of the characterized tyrosinases this range is considerably more narrow, e.g. for the heterologously expressed tyrosinase from Polyporus arcularius it is found between pH 5–649 and the homologously overexpressed T. reesei TYR2 was found to be almost fully active in the range of pH 6–9.545.
The kinetic characterization of activated AbPPO4 shows low specificity and high reaction rates for the tested substrates, especially the diphenols (see Table 2). Kinetic parameters of recombinant fungal tyrosinases on L-tyrosine were reported for AbPPO2 produced in Saccharomyces cerevisiae (Km = 0.302 µM, kcat = 11.39 s−1)58 and for MelB from Aspergillus oryzae produced in E. coli (Km = 43 µM, kcat = 49 s−1)59, making these enzymes both more specific towards L-tyrosine as well as faster on this substrate than activated AbPPO4. For TYR1 from P. nameko the kinetic parameters on tyrosine could not be determined due to insufficient solubility of the substrate52. For L-DOPA a slightly higher number of values (enzyme name: Km in µM | kcat in s−1) are reported for the recombinant tyrosinases from A. bisporus (AbPPO2: 1.22 | 141)58, P. nameko (TYR1: 1930 | 478)52, P. arcularius (Photo-regulated tyrosinase: 1040 | 223)49 and T. reesei (TYR2: 3000 | 22)45. In comparison to those enzymes activated AbPPO4 is less specific for L-DOPA and much faster on this substrate.
Conclusions
In conclusion, a protocol for the production of mushroom tyrosinase was established which is able to produce both latent and active tyrosinase in a pure form. Using this protocol it is possible to provide quantities of mushroom tyrosinase sufficient for providing even larger research projects with a defined tyrosinase preparation that does not suffer from the isoenzyme mixture and the side-activities frequently present in commercial preparations of tyrosinase isolated from mushrooms.
Methods
If not indicated otherwise the chemicals used were purchased from Sigma-Aldrich (Vienna, Austria) or Carl Roth (Karlsruhe, Germany) and were at least of analytical grade. The methyl esters of L- and D-tyrosine were synthesized at the Department of Biophysical Chemistry, University of Vienna and are characterized by 1H-NMR and ESI-MS (see supporting information and Figures S1 and S2).
RNA-extraction and cDNA-synthesis
RNA extraction and cDNA synthesis have been performed according to standard procedures60 and are described in detail in the SI.
Cloning of the AbPPO4 gene
The gene encoding AbPPO4 was cloned out of frame into the expression vector pGEX-6P-1 (GE Healthcare Europe; Freiburg, Germany) and was brought in frame by PCR-based mutagenesis (for details see SI).
Mutagenesis of AbPPO4
As the enzyme isolated from the natural source did not contain the last 46 amino acids that are encoded by its gene27, the cloned gene was adjusted accordingly. Towards that end the base triplets encoding the respective amino acids were deleted from the gene using the Q5® Site-Directed Mutagenesis Kit (NEB). Removal of the sequence corresponding to amino acids A566 to F611 was accomplished with the two primers lAbPPO4_fwd and lAbPPO4_rev, while E384 to F611 was removed using lAbPPO4_fwd and aAbPPO4_rev resulting in the bases encoding just the main domain of the tyrosinase27, 33.
Expression of AbPPO4
AbPPO4 was expressed in E. coli BL21(DE3) in auto-inducing medium, namely ZYM-5052 without the trace element solution61 but with an additional 500 mM of NaCl. Cultures were grown at 20 °C in shaking flasks in media containing 100 mg l−1 Na-Ampicillin for 20 h. Then, 0.5 mM copper sulfate was added and expression was continued for 20 more hours (for details see SI).
Isolation and purification of the recombinant tyrosinase
The pelleted cells were washed with 10% of the original culture volume of 9 g l−1 NaCl in ddH2O, repelleted by centrifugation (8 min @ 3000 × g and 4 °C) and resuspended in lysis buffer (25 mM HEPES, 150 mM NaCl, 5 mM Na2MgEDTA set to pH 7.3 with NaOH with the following three components being added immediately before use: 2 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 100 mg l−1 hen egg white lysozyme) at 100 g of wet cells per l. Cells were disrupted by 2 passages through a french press62 at a cell pressure of 850 bar. The samples were cooled on ice in between the passages through the french press and fresh PMSF was added to a final concentration of 2 mM in two portions at the end of each passage. The lysate was cleared by centrifugation (15 min @ 30800 × g and 4 °C), filtered (0.45 µm pore size PES membrane) and applied to a 5 ml GSTrap FF column at a flow rate of 0.5 ml min−1. The column was kept at 4 °C and 50 mM Tris-HCl, 150 mM NaCl pH 8.0 (@ 4 °C) was used as the mobile phase for elution of unbound material while the same solution with 20 mM reduced glutathione served as the elution buffer. The eluted fractions containing the fusion protein were concentrated by ultrafiltration (Vivaspin® 20, 30 kDa molecular weight cut-off) which was also applied for buffer exchange.
GST was cleaved from the fusion protein by the action of picornain 3C (human rhinovirus serotype 14 protease 3C, HRV 3C) which was applied as a fusion-protein with GST (production protocol in the SI). 1 µg of protease was applied per 150 µg of GST-AbPPO4 fusion protein and the proteolysis reaction was allowed to proceed for at least 18 h at 4 °C in the running buffer of the affinity chromatography supplemented with 1 mM DTT in order to preserve the activity of the cysteine protease HRV 3C.
After enzymatic cleavage, the tyrosinase was separated from the fusion partner as well as the protease by a second passage through the affinity column using identical conditions as for the first chromatographic step. The column flow-through containing the protein of interest was concentrated by ultrafiltration (Vivaspin® 20, 30 kDa molecular weight cut-off) and its buffer was exchanged to 10 mM HEPES pH 7.5 (@ 4 °C). This preparation was diluted to a concentration of 20 g l−1 and stored at 4 °C until use.
Enzymatic activity assay
Tyrosinase activity was routinely assayed on 1 mM L-tyrosine in 50 mM sodium citrate buffer pH 6.8 at 25 °C. For activation of the latent enzyme 2 mM SDS were included in the assay mixture27. The monitored species was dopachrome at 475 nm (ε475 = 3600 M−1 cm−1)63, volumetric enzymatic activities were calculated from the linear part of the absorption-time curves (after the lag-phase but before the subsequent reactions towards melanin contribute significantly). One unit of enzymatic activity (U) was defined as the amount of enzyme that catalyzes the conversion of 1 µmol of substrate per minute of reaction.
Intact protein mass spectrometry
For ESI-MS, which was done at the Mass Spectrometry Centre at the University of Vienna, the buffer of the protein solutions was exchanged to 5 mM ammonium hydrogen carbonate pH 7.8 by repeated ultrafiltration (Vivaspin® 500, 30 kDa molecular weight cut-off). For introduction into the nanoESI-QTOF mass spectrometer (maXis 4 G UHR-TOF from Bruker, Billerica, MA, USA; providing a mass accuracy better than 5 ppm) by a syringe pump (KDS 100 from KD Scientific, Holliston, MA, USA) @ 3 µl min−1 the protein solutions were diluted to approximately 1 µM in an aqueous solution containing 2% (v/v) acetonitrile and 1‰ (v/v) formic acid.
Enzyme kinetics
A spectrophotometric assay detecting the appearance of the product in the reaction solution was applied for the determination of the kinetic parameters of AbPPO4. Since the o-quinones generated by the enzymatic action of tyrosinase do not give rise to a stable and soluble product they were trapped by the potent nucleophile 3-methyl-2-benzothiazolinone hydrazone (MBTH). MBTH couples to o-quinones via its amino group generating reasonably stable adducts that remain soluble and are easily detected photometrically64. Absorption curves and spectra were recorded on a Shimadzu UV-1800 spectrophotometer applying 1 cm cuvettes which were kept at 25 °C by a Julabo F25 MH thermostat in a circulating water-bath. Kinetic measurements were done in a total volume of 1 ml containing 50 mM sodium citrate buffer pH 6.8, 5 mM MBTH, 2% (v/v) N,N-dimethylformamide and different concentrations of the substrates to be tested as well as AbPPO4 (0.23 to 46 nM).
Molar absorption coefficients were determined from rapid oxidation of small concentrations of substrate under standard assay conditions applying tyrosinase concentrations in the µM range. Km and kcat, the two parameters of the Michaelis-Menten model65, were calculated from the steady-state rate of product formation for the different substrate concentrations tested. Measurements were performed in triplicate and the reciprocals of the variances of the observed slopes were used as weights for the nonlinear regression applying the Levenberg-Marquardt algorithm66 as implemented in the program Dataplot (version 11/2010). Initial estimates for the two free parameters were generated by applying the Hanes-Woolf linearization of the Michaelis-Menten equation67.
Proteolytic activation of AbPPO4
Latent AbPPO4 was converted into its active form by treatment with proteinase K. The protease was used at a ratio of 1:10 (equivalent to 45 µg of proteinase K per mg of latent AbPPO4) in a reaction buffer containing 50 mM Tris-HCl pH 8 @ 25 °C, 100 mM sodium ascorbate and 20 g l−1 of latent AbPPO4 for a total reaction time of 90 min. The reaction was stopped by addition of 2 mM PMSF after which the solution was concentrated to less than 70 µl by ultrafiltration (Vivaspin® 500, 30 kDa molecular weight cut-off) and applied onto a size exclusion column (Superdex 200 Increase from GE Healthcare) equilibrated with 50 mM sodium citrate pH 6.8 and run with the same buffer at 4 °C and with a flow rate of 0.5 ml min−1. The eluted fractions possessing tyrosinase activity were pooled and concentrated by ultrafiltration (Vivaspin® 500, 30 kDa molecular weight cut-off).
Protein crystallization, X-ray diffraction and model building
Conditions for the growth of AbPPO4 crystals were refined based on an initial hit obtained with sodium cacodylate and PEG 4000 in a hanging drop vapour diffusion setup. Single crystals suitable for crystallography grew over the course of a few days in hanging drops initially made up of 1 µl of a 10 g l−1 solution of latent AbPPO4 in 10 mM HEPES pH 7.5 mixed with 1 µl of reservoir solution containing 50 mM sodium cacodylate pH 5.8 and 13% (w/w) PEG 4000 which was equilibrated via vapour diffusion with 1 ml of reservoir solution at 293 K. Crystals were harvested using Kapton® loops (Hampton Research, Aliso Viejo, CA, USA), soaked with cryo-protectant (50 mM sodium cacodylate pH 5.8 and 40% (w/w) PEG 4000) and plunged into liquid nitrogen where they remained until the diffraction experiment.
Data were collected at beamline ID-23 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) at 100 K applying a wavelength of 0.873 Å (14.2 keV) and a PILATUS2 3 M detector. Data reduction (for details see SI) was carried out using XDS68. Final model quality was evaluated by the MolProbity server69 and the model has been deposited in the PDB under entry number 5M6B.
References
Claus, H. & Decker, H. Bacterial tyrosinases. Syst. Appl. Microbiol. 29, 3–14, doi:10.1016/j.syapm.2005.07.012 (2006).
Lerner, A. B. Effect of Ions on Melanin Formation. J. Invest. Dermatol. 18, 47–52, doi:10.1038/jid.1952.6 (1952).
Mason, H. S. The Chemistry of Melanin: III. Mechanism of the Oxidation of Dihydroxyphenylalanine by Tyrosinase. J. Biol. Chem. 172, 83–99 (1948).
Rodríguez-López, J. N., Tudela, J., Varón, R., García-Carmona, F. & García-Cánovas, F. Analysis of a kinetic model for melanin biosynthesis pathway. J. Biol. Chem. 267, 3801–3810 (1992).
Pfiffner, E. & Lerch, K. Histidine at the active site of Neurospora tyrosinase. Biochemistry 20, 6029–6035, doi:10.1021/bi00524a017 (1981).
Hazes, B. et al. Crystal structure of deoxygenated Limulus polyphemus subunit II hemocyanin at 2.18A resolution: clues for a mechanism for allosteric regulation. Protein Sci. 2, 597–619, doi:10.1002/pro.5560020411 (1993).
Kitajima, N., Fujisawa, K., Moro-oka, Y. & Toriumi, K. µ-η2:η2-Peroxo binuclear copper complex, [Cu(HB(3,5-(Me2CH)2pz)3)]2(O2). J. Am. Chem. Soc. 111, 8975–8976, doi:10.1021/ja00206a062 (1989).
Rompel, A. et al. Spectroscopic and EXAFS studies on catechol oxidases with dinuclear copper centers of type 3: Evidence for μ-η2:η2-peroxo-intermediates during the reaction with catechol. J. Inorg. Biochem. 59, 715, doi:10.1016/0162-0134(95)97803-X (1995).
Rompel, A. et al. Purification and spectroscopic studies on catechol oxidases from Lycopus europaeus and Populus nigra: Evidence for a dinuclear copper center of type 3 and spectroscopic similarities to tyrosinase and hemocyanin. JBIC J. Biol. Inorg. Chem. 4, 56–63, doi:10.1007/s007750050289 (1999).
Ito, S. A Chemist’s View of Melanogenesis. Pigment Cell Res 16, 230–236, doi:10.1034/j.1600-0749.2003.00037.x (2003).
Hearing, V. J. & Tsukamoto, K. Enzymatic control of pigmentation in mammals. FASEB J. 5, 2902–2909 (1991).
Mason, H. S. & Wright, C. I. The chemistry of melanin v. oxidation of dihydroxyphenylalanine by tyrosinase. J. Biol. Chem. 180, 235–247 (1949).
Pawelek, J. M. After Dopachrome? Pigment Cell Res. 4, 53–62, doi:10.1111/j.1600-0749.1991.tb00315.x (1991).
Polacheck, I. & Kwon-Chung, K. J. Melanogenesis in Cryptococcus neoformans. J. Gen. Microbiol. 134, 1037–1041, doi:10.1099/00221287-134-4-1037 (1988).
Burzio, L. A. & Waite, J. H. Reactivity of peptidyl-tyrosine to hydroxylation and cross-linking. Protein Sci. 10, 735–740, doi:10.1110/ps.44201 (2001).
Ito, S., Kato, T., Shinpo, K. & Fujita, K. Oxidation of tyrosine residues in proteins by tyrosinase. Formation of protein-bonded 3,4-dihydroxyphenylalanine and 5-S-cysteinyl-3,4-dihydroxyphenylalanine. Biochem. J. 222, 407–411, doi:10.1042/bj2220407 (1984).
Burzio, L. A. & Waite, J. H. Cross-Linking in Adhesive Quinoproteins: Studies with Model Decapeptides. Biochemistry 39, 11147–11153, doi:10.1021/bi0002434 (2000).
McDowell, L. M., Burzio, L. A., Waite, J. H. & Schaefer, J. Rotational Echo Double Resonance Detection of Cross-links Formed in Mussel Byssus under High-Flow Stress. J. Biol. Chem. 274, 20293–20295, doi:10.1074/jbc.274.29.20293 (1999).
Faccio, G., Kruus, K., Saloheimo, M. & Thöny-Meyer, L. Bacterial tyrosinases and their applications. Process Biochem. 47, 1749–1760, doi:10.1016/j.procbio.2012.08.018 (2012).
Pretzler, M., Bijelic, A., Rompel, A. Fungal Tyrosinases: Why Mushrooms Turn Brown. In: Ref. Module Chem. Mol. Sci. Chem. Eng. Elsevier, doi:10.1016/B978-0-12-409547-2.11521-5 (2015).
Seo, S.-Y., Sharma, V. K. & Sharma, N. Mushroom Tyrosinase: Recent Prospects. J. Agric. Food Chem. 51, 2837–2853, doi:10.1021/jf020826f (2003).
Flurkey, A. et al. Enzyme, Protein, Carbohydrate, and Phenolic Contaminants in Commercial Tyrosinase Preparations: Potential Problems Affecting Tyrosinase Activity and Inhibition Studies. J. Agric. Food Chem. 56, 4760–4768, doi:10.1021/jf800109a (2008).
Rescigno, A., Zucca, P., Flurkey, A., Inlow, J. & Flurkey, W. H. Identification and discrimination between some contaminant enzyme activities in commercial preparations of mushroom tyrosinase. Enzyme Microb. Technol. 41, 620–627, doi:10.1016/j.enzmictec.2007.05.009 (2007).
Morin, E. et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci USA 109, 17501–17506, doi:10.1073/pnas.1206847109 (2012).
Weijn, A., Bastiaan-Net, S., Wichers, H. J. & Mes, J. J. Melanin biosynthesis pathway in Agaricus bisporus mushrooms. Fungal Genet. Biol. 55, 42–53, doi:10.1016/j.fgb.2012.10.004 (2013).
Ismaya, W. T. et al. Crystal Structure of Agaricus bisporus Mushroom Tyrosinase: Identity of the Tetramer Subunits and Interaction with Tropolone. Biochemistry 50, 5477–5486, doi:10.1021/bi200395t (2011).
Mauracher, S. G. et al. High level protein-purification allows the unambiguous polypeptide determination of latent isoform PPO4 of mushroom tyrosinase. Phytochemistry 99, 14–25, doi:10.1016/j.phytochem.2013.12.016 (2014).
Wu, J. et al. Cloning, characterization and expression of two new polyphenol oxidase cDNAs from Agaricus bisporus. Biotechnol. Lett. 32, 1439–1447, doi:10.1007/s10529-010-0329-2 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081, doi:10.1038/nprot.2009.86 (2009).
Smith, D. B. & Johnson, K. S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40, doi:10.1016/0378-1119(88)90005-4 (1988).
Dirks-Hofmeister, M. E., Kolkenbrock, S. & Moerschbacher, B. M. Parameters That Enhance the Bacterial Expression of Active Plant Polyphenol Oxidases. PLoS ONE 8, e77291, doi:10.1371/journal.pone.0077291 (2013).
Deng, J. et al. An improved protocol for rapid freezing of protein samples for long-term storage. Acta Crystallogr. Sect. D 60, 203–204, doi:10.1107/S0907444903024491 (2004).
Mauracher, S. G., Molitor, C., Al-Oweini, R., Kortz, U. & Rompel, A. Latent and active abPPO4 mushroom tyrosinase cocrystallized with hexatungstotellurate(VI) in a single crystal. Acta Crystallogr. Sect. D 70, 2301–2315, doi:10.1107/S1399004714013777 (2014).
Steill, J. D., Szczepanski, J., Oomens, J., Eyler, J. R. & Brajter-Toth, A. Structural characterization by infrared multiple photon dissociation spectroscopy of protonated gas-phase ions obtained by electrospray ionization of cysteine and dopamine. Anal. Bioanal. Chem. 399, 2463–2473, doi:10.1007/s00216-010-4582-y (2011).
Bijelic, A. & Rompel, A. The use of polyoxometalates in protein crystallography–An attempt to widen a well-known bottleneck. Coord. Chem. Rev. 299, 22–38, doi:10.1016/j.ccr.2015.03.018 (2015).
Mauracher, S. G., Molitor, C., Al-Oweini, R., Kortz, U. & Rompel, A. Crystallization and preliminary X-ray crystallographic analysis of latent isoform PPO4 mushroom (Agaricus bisporus) tyrosinase. Acta Crystallogr. Sect. F 70, 263–266, doi:10.1107/S2053230X14000582 (2014).
Molitor, C., Bijelic, A. & Rompel, A. In situ formation of the first proteinogenically functionalized [TeW6O24O2(Glu)]7− structure reveals unprecedented chemical and geometrical features of the Anderson-type cluster. Chem. Commun. 52, 12286–12289, doi:10.1039/c6cc07004c (2016).
Land, E. J., Ramsden, C. A. & Riley, P. A. The Mechanism of Suicide-Inactivation of Tyrosinase: A Substrate Structure Investigation. Tohoku J. Exp. Med. 212, 341–348, doi:10.1620/tjem.212.341 (2007).
Muñoz-Muñoz, J. L. et al. Unravelling the suicide inactivation of tyrosinase: A discrimination between mechanisms. J. Mol. Catal. B Enzym 75, 11–19, doi:10.1016/j.molcatb.2011.11.001 (2012).
Molitor, C. et al. Latent and active aurone synthase from petals of C. grandiflora: a polyphenol oxidase with unique characteristics. Planta 242, 519–537, doi:10.1007/s00425-015-2261-0 (2015).
Ren, Q., Henes, B., Fairhead, M. & Thony-Meyer, L. High level production of tyrosinase in recombinant Escherichia coli. BMC Biotechnol. 13, 18, doi:10.1186/1472-6750-13-18 (2013).
Lai, X., Soler-Lopez, M., Wichers, H. J. & Dijkstra, B. W. Large-Scale Recombinant Expression and Purification of Human Tyrosinase Suitable for Structural Studies. PLoS ONE 11, e0161697, doi:10.1371/journal.pone.0161697 (2016).
Kaintz, C. et al. Cloning and functional expression in E. coli of a polyphenol oxidase transcript from Coreopsis grandiflora involved in aurone formation. FEBS Lett 588, 3417–3426, doi:10.1016/j.febslet.2014.07.034 (2014).
Halaouli, S. et al. Cloning and characterization of a tyrosinase gene from the white-rot fungus Pycnoporus sanguineus, and overproduction of the recombinant protein in Aspergillus niger. Appl. Microbiol. Biotechnol. 70, 580–589, doi:10.1007/s00253-005-0109-4 (2006).
Selinheimo, E. et al. Production and characterization of a secreted, C-terminally processed tyrosinase from the filamentous fungus Trichoderma reesei. FEBS J. 273, 4322–4335, doi:10.1111/j.1742-4658.2006.05429.x (2006).
Westerholm-Parvinen, A. et al. Expression of the Trichoderma reesei tyrosinase 2 in Pichia pastoris: Isotopic labeling and physicochemical characterization. Protein Expr. Purif. 55, 147–158, doi:10.1016/j.pep.2007.04.014 (2007).
Faccio, G., Arvas, M., Thöny-Meyer, L. & Saloheimo, M. Experimental and bioinformatic investigation of the proteolytic degradation of the C-terminal domain of a fungal tyrosinase. J. Inorg. Biochem. 121, 37–45, doi:10.1016/j.jinorgbio.2012.12.006 (2013).
Kawamura-Konishi, Y., Maekawa, S., Tsuji, M. & Goto, H. C-terminal processing of tyrosinase is responsible for activation of Pholiota microspora proenzyme. Appl. Microbiol. Biotechnol. 90, 227–234, doi:10.1007/s00253-010-3039-8 (2011).
Marková, E. et al. Recombinant Tyrosinase from Polyporus arcularius: Overproduction in Escherichia coli, Characterization and Use in a Study of Aurones as Tyrosinase Effectors. J. Agric. Food Chem. 64, 2925–2931, doi:10.1021/acs.jafc.6b00286 (2016).
Flurkey, W. H. & Inlow, J. K. Proteolytic processing of polyphenol oxidase from plants and fungi. J. Inorg. Biochem. 102, 2160–2170, doi:10.1016/j.jinorgbio.2008.08.007 (2008).
Lerch, K. Amino acid sequence of tyrosinase from Neurospora crassa. Proc. Natl. Acad. Sci USA 75, 3635–3639, doi:10.1073/pnas.75.8.3635 (1978).
Kawamura-Konishi, Y. et al. Purification, Characterization, and Molecular Cloning of Tyrosinase from Pholiota nameko. Biosci. Biotechnol. Biochem. 71, 1752–1760, doi:10.1271/bbb.70171 (2007).
Marusek, C. M., Trobaugh, N. M., Flurkey, W. H. & Inlow, J. K. Comparative analysis of polyphenol oxidase from plant and fungal species. J. Inorg. Biochem. 100, 108–123, doi:10.1016/j.jinorgbio.2005.10.008 (2006).
Yurkow, E. J. & Laskin, J. D. Purification of tyrosinase to homogeneity based on its resistance to sodium dodecyl sulfate-proteinase K digestion. Arch. Biochem. Biophys. 275, 122–129, doi:10.1016/0003-9861(89)90356-1 (1989).
Marqués, L., Fleuriet, A., Cleyet-Marel, J.-C. & Macheix, J.-J. Purification of an apple polyphenol oxidase isoform resistant to SDS-proteinase K digestion. Phytochemistry 36, 1117–1121, doi:10.1016/S0031-9422(00)89623-5 (1994).
Fujita, Y., Uraga, Y. & Ichisima, E. Molecular cloning and nucleotide sequence of the protyrosinase gene, melO, from Aspergillus oryzae and expression of the gene in yeast cells. Biochim. Biophys. Acta BBA. Gene Struct. Expr 1261, 151–154, doi:10.1016/0167-4781(95)00011-5 (1995).
Espín, J. C. & Wichers, H. J. Activation of a Latent Mushroom (Agaricus bisporus) Tyrosinase Isoform by Sodium Dodecyl Sulfate (SDS). Kinetic Properties of the SDS-Activated Isoform. J. Agric. Food Chem. 47, 3518–3525, doi:10.1021/jf981275p (1999).
Lezzi, C., Bleve, G., Spagnolo, S., Perrotta, C. & Grieco, F. Production of recombinant Agaricus bisporus tyrosinase in Saccharomyces cerevisiae cells. J. Ind. Microbiol. Biotechnol. 39, 1875–1880, doi:10.1007/s10295-012-1192-z (2012).
Fujieda, N. et al. Multifunctions of MelB, a Fungal Tyrosinase from Aspergillus oryzae. ChemBioChem 13, 193–201, doi:10.1002/cbic.201100609 (2012).
Mülhardt, C. Der Experimentator Molekularbiologie/Genomics. In: Mülhardt, C., editor. Springer Spektrum. Experimentator 7, pp. 140–142 (2013).
Studier, F. W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234, doi:10.1016/j.pep.2005.01.016 (2005).
French, C. S., Milner, H. W. [9] Disintegration of bacteria and small particles by high-pressure extrusion. In: Academic Press. Methods in Enzymology, Vol. 1, pp. 64–67 (1955).
Gandía-Herrero, F., Jiménez-Atiénzar, M., Cabanes, J., García-Carmona, F. & Escribano, J. Differential Activation of a Latent Polyphenol Oxidase Mediated by Sodium Dodecyl Sulfate. J. Agric. Food Chem. 53, 6825–6830, doi:10.1021/jf050505e (2005).
Winder, A. J. & Harris, H. New assays for the tyrosine hydroxylase and dopa oxidase activities of tyrosinase. Eur. J. Biochem. 198, 317–326, doi:10.1111/j.1432-1033.1991.tb16018.x (1991).
Michaelis, L. & Menten, M. L. Die Kinetik der Invertinwirkung. Biochem. Z. 49, 333–369 (1913).
Marquardt, D. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 11, 431–441, doi:10.1137/0111030 (1963).
Hanes, C. S. CLXVII. Studies on plant amylases I. The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 26, 1406–1421, doi:10.1042/bj0261406 (1932).
Kabsch, W. XDS. Acta Crystallogr. Sect. D 66, 125–132, doi:10.1107/S0907444909047337 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21, doi:10.1107/S0907444909042073 (2010).
Bijelic, A., Pretzler, M., Molitor, C., Zekiri, F. & Rompel, A. The Structure of a Plant Tyrosinase from Walnut Leaves Reveals the Importance of “Substrate-Guiding Residues” for Enzymatic Specificity. Angew. Chem. Int. Ed. 54, 14677–14680, doi:10.1002/anie.201506994 (2015); Angew. Chem. 127, 14889–14893, doi:10.1002/ange.201506994 (2015).
Acknowledgements
The research was funded by the Austrian Science Fund (FWF): P25217. We thank the beamline scientists Dr. Anja Burkhardt (DESY P11, I-20120633 EC) and Dr. Ulrich Zander (ESRF ID23-2, mx1616) for their generous support during the allocated beam-times. The authors wish to thank Emir Al-Sayed, MSc for the synthesis and characterization of D- and L-tyrosine methylester and Ioannis Kampatsikas, M.Sc. for valuable discussions concerning this work.
Author information
Authors and Affiliations
Contributions
M.P., A.B. and A.R. jointly designed the experiments, M.P. performed the experiments and analysed the data and wrote the paper with input from all co-authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pretzler, M., Bijelic, A. & Rompel, A. Heterologous expression and characterization of functional mushroom tyrosinase (AbPPO4). Sci Rep 7, 1810 (2017). https://doi.org/10.1038/s41598-017-01813-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01813-1
This article is cited by
-
Tissue engineering modalities in skeletal muscles: focus on angiogenesis and immunomodulation properties
Stem Cell Research & Therapy (2023)
-
Biochemical characterization of Dimocarpus longan polyphenol oxidase provides insights into its catalytic efficiency
Scientific Reports (2022)
-
Tyrosinase and laccase-producing Bacillus aryabhattai TFG5 and its role in the polymerization of phenols
BMC Microbiology (2021)
-
Wells–Dawson phosphotungstates as mushroom tyrosinase inhibitors: a speciation study
Scientific Reports (2021)
-
Polyphenol oxidases exhibit promiscuous proteolytic activity
Communications Chemistry (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
