
Abstract
Duck enteritis virus (DEV) UL54 is a homologue of human herpes simplex virus-1 (HSV-1) ICP27, which plays essential regulatory roles during infection. Our previous studies indicated that DEV UL54 is an immediate-early protein that can shuttle between the nucleus and the cytoplasm. In the present study, we found that UL54-deleted DEV (DEV-ΔUL54) exhibits growth kinetics, a plaque size and a viral DNA copy number that are significantly different from those of its parent wild-type virus (DEV-LoxP) and the revertant (DEV-ΔUL54 (Revertant)). Relative viral mRNA levels, reflecting gene expression, the transcription phase and the translation stage, are also significantly different between DEV-ΔUL54-infected cells and DEV-LoxP/DEV-ΔUL54 (Revertant)-infected cells. However, the localization pattern of UL30 mRNA is obviously changed in DEV-ΔUL54-infected cells. These findings suggest that DEV UL54 is important for virus growth and may regulate viral gene expression during transcription, mRNA export and translation.
Similar content being viewed by others


Introduction
Duck enteritis virus (DEV), also known as duck plague virus (DPV), is an extensively studied alpha-herpesvirus. This virus is the causative agent of duck virus enteritis (DVE), an acute haemorrhagic disease that causes significant economic losses in waterfowl-based industry due to high mortality and low egg-laying rates. The DEV genome is a linear, double-stranded DNA that is divided into a unique long region (UL) and a unique short region (US) flanked by a short internal repeat sequence (IRS) and a short terminal repeat sequence (TRS)1. The sequence of the complete DEV genome and the functions of several viral genes, except UL54, have been reported, which could help in eliminating DEV completely2,3,4.
As a conserved protein5, herpes simplex virus-1 (HSV-1) ICP27, a homologue of UL54, is required for viral replication6. This protein possesses a shuttling property with nuclear and cytoplasmic activities; as a result, ICP27 is multi- functional, playing roles in both the positive and the negative regulation of expression of different target genes7,8,9,10. In the nucleus, ICP27 can stimulate the transcription11,12,13,14,15 of early and late viral genes, affecting pre-mRNA splicing16,17,18,19,20. During dynamic shuttling, ICP27 can promote the export of viral intronless mRNAs, which are less efficiently exported than spliced RNAs21,22,23,24,25,26,27. The most important effect of this protein’s cytoplasmic activities is stimulation of the translation of certain viral transcripts28,29,30. Additionally, ICP27 has been identified to promote genomic DNA replication, which also occurs in the nucleus31. These findings show that HSV-1 ICP27 is important for modulating the biogenesis of DNA and mRNA, which is necessary for virus growth.
To date, there have been few reports of the role of UL54 in other herpesviruses and even fewer reports examining UL54 in DEV. In a previous study, simplified bioinformatics analyses of DEV UL54 were first conducted to lay a theoretical basis32. After that, DEV UL54 was expressed as a fusion protein, and in this context, a specific anti-UL54 antibody was evaluated. The intracellular localization and levels of the DEV UL54 transcript and protein were then studied, and the results showed that DEV UL54 is a nuclear protein that is expressed as early as 0.5 h after infection, with a peak at 24 h. According to a pharmacological inhibition test, UL54 was confirmed to be an immediate-early gene based on its insensitivity to the DNA polymerase inhibitor ganciclovir (GCV) and the protein synthesis inhibitor cycloheximide (CHX). Later, the DEV UL54 protein was identified to shuttle between the nucleus and the cytoplasm, and the predicted nuclear localization sequences (NLSs) and nuclear export signal (NES)33, 34 were evaluated. The findings suggested that DEV UL54 also plays vital regulatory roles in a similar way to HSV-1 ICP27.
In the present study, we first identified and characterized DEV-ΔUL54 and DEV-ΔUL54 (Revertant) constructs34. Based on results regarding the growth curve, plaque area and viral genomic DNA copy number, we found that DEV UL54 is important for virus growth. Then the viral mRNA levels, and particularly the total RNA, nuclear RNA and ribosome-nascent chain complex (RNC)-containing RNA levels, were then analysed by real-time PCR to determine the effects of DEV UL54 on viral gene expression, transcription and translation. Furthermore, the localization of UL30 mRNA in DEV-ΔUL54, DEV-ΔUL54 (Revertant) and DEV-LoxP was examined by fluorescence in situ hybridization (FISH). The results showed that DEV UL54 could inhibit or enhance viral gene expression, transcription and translation and promote the export of UL30 mRNA. Our results thus help to address a gap in the field of research on DEV UL54 function.
Results
DEV UL54 is important for viral replication
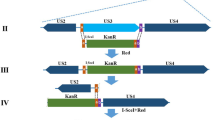
To investigate the functional roles of DEV UL54, we first constructed DEV CHv-BAC-ΔUL54 and DEV CHv-BAC-ΔUL54 (Revertant) by genetic manipulation of DEV CHv-BAC-G34 (Fig. 1A, lower panels). With the help of the Cre-LoxP system, we obtained DEV-ΔUL54 and DEV-ΔUL54 (Revertant) after removing the EGFP-BAC tag (Fig. 1A, upper panels). DEV-ΔUL54 and DEV-ΔUL54 (Revertant) were identified by PCR (Fig. 1B), indirect immunofluorescence assay (IFA) (Fig. 1C) and Western blotting (Fig. 1D). The PCR results showed that the target fragments of DEV CHv-BAC-ΔUL54/DEV CHv-BAC-ΔUL54 (Revertant) and DEV-ΔUL54/DEV-ΔUL54 (Revertant) were approximately 10000 bp and 1700 bp, respectively, in size. In the IFA and Western blot analyses, no specific green fluorescence or specific band was observed for DEV-ΔUL54, which was not the case for DEV-ΔUL54 (Revertant). DEV UL13 was chosen as a control to show that the cells were successfully infected. Taken together, these results implied that the DEV UL54 deletion (DEV-ΔUL54) and revertant (DEV-ΔUL54 (Revertant)) were successfully constructed.

Construction of DEV-ΔUL54 and DEV-ΔUL54 (Revertant). (A) Construction of DEV-ΔUL54 and DEV-ΔUL54 (Revertant). The DEV CHv-BAC-ΔUL54 and DEV CHv-BAC-ΔUL54 (Revertant) were constructed with the Red recombinant system and rescued in DEFs cells. Using the Cre-LoxP system, the BAC-EGFP tag was removed. (B) Identification of DEV-ΔUL54 and DEV-ΔUL54 (Revertant) by PCR. M: DL15000; 1: DEV CHv-BAC-ΔUL54; 2: DEV-ΔUL54; 3: DEV-ΔUL54 (Revertant); 4: DEV CHv-BAC-ΔUL54 (Revertant). The target fragment size in DEV CHv-BAC-ΔUL54/DEV CHv-BAC-ΔUL54 (Revertant) was approximately 10000 bp, and it was nearly 1700 bp for DEV-ΔUL54/DEV-ΔUL54 (Revertant). (C) Identification of DEV-ΔUL54 and DEV-ΔUL54 (Revertant) by IFA. DEFs cells that were infected with DEV-ΔUL54 or DEV-ΔUL54 (Revertant) were subjected to IFA with Anti-UL54 polyclonal antibody as a primary antibody; DEV-ΔUL54 treated with Anti-UL13 polyclonal antibody as primary antibody was used as a control. (D) Identification of DEV-ΔUL54 and DEV-ΔUL54 (Revertant) by Western blot. Proteins of DEFs cells infected with DEV-ΔUL54 or DEV-ΔUL54 (Revertant) were subjected to Western blot with Anti-UL54 polyclonal antibody as a primary antibody. 1: DEV-ΔUL54 (Revertant); 2: DEV-ΔUL54; 3: DEV-ΔUL54 subjected with Anti-UL13 polyclonal antibody as a primary antibody.
Analyses of the growth curve and a plaque assay revealed that DEV-ΔUL54 could efficiently grow in duck embryo fibroblasts (DEFs) (Fig. 2A) while producing a smaller plaque size (Fig. 2B,C). Obvious recovery of DEV-ΔUL54 (Revertant) compared with DEV-ΔUL54 was found, suggesting that the defects resulted from the lack of the UL54 gene.

Growth curve and plaque size of the DEV-derived viruses. (A) Growth curve. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at a titer of 200 TCID50 and harvested at different time points. The curve was generated based on the titers of different harvests by testing TCID50. a: p < 0.05. (B) Phenotypes of viral plaques. Plaques were formed on DEFs under a semi-solid medium overlay containing methylcellulose. After fixation, crystal violet staining was performed. (C) Analysis of the plaque area. The mean areas of plaque were analyzed using IPP 6.0. **p < 0.01.
Additionally, real-time PCR was performed, and the viral DNA copy number was estimated at various time points using the fitted equation of the standard curve (Fig. 3A): Y = −3.323X-49.610 (E = 100.0%, R^2 = 0.999). Compared with DEV-LoxP and DEV-ΔUL54 (Revertant), a notable decrease was observed in DEV-ΔUL54 (Fig. 3B), coinciding with the results of the growth curve and plaque assay analyses.

Viral DNA copy number of the DEV-derived viruses. (A) Construction of the standard curve. (B) Levels of DEV-derived genomic DNA. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at 200 TCID50. At different time points, total viral DNA was purified, and quantitative PCR was performed. **p < 0.01.
DEV UL54 regulates viral gene expression during transcription and translation
Next, the effects of DEV UL54 on viral mRNA expression were studied. The mRNA levels of viral genes, including UL19, UL30, UL48, gC, gD, and gK were estimated by relative real-time PCR. As shown in Fig. 4, DEV UL54 continuously promoted the viral mRNA expression of UL30 and gC, and an increase was observed for UL48 and gD, except in the early stage of infection. UL19 could be both positively and negatively regulated by UL54, although the negative activity was dominant. Finally, UL54 could enhance or repress gK expression in early infection. Considering these results together, we concluded that the DEV UL54 gene could inhibit or augment viral mRNA expression.

Effects of DEV UL54 on viral mRNA expression. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at 200 TCID50, and RT-PCR was performed to assess the relative expression levels of UL19, UL30, UL48, gC, gD and gK at different time points. *p < 0.05; **p < 0.01.
The transcriptional mRNA levels of UL19, UL30, UL48, gC, gD, and gK were then analysed by RT-PCR after performing a nuclear run-off assay. As shown in Fig. 5, the mRNA transcription levels for all candidate genes were significantly lower in DEV-ΔUL54 than in DPV-LoxP/DEV-ΔUL54 (Revertant), except for UL30 at 12 h. This result indicated that DEV UL54 predominantly promoted viral gene transcription during infection.

Regulation of DEV UL54 on viral gene transcription. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at 200 TCID50 and the nuclei were extracted. After a nuclear run-off assay, RT-PCR was performed to assess the relative transcriptional mRNA levels of UL19, UL30, UL48, gC, gD and gK at different time points. *p < 0.05; **p < 0.01.
As shown in Fig. 6, the mRNA translation levels for target genes were significantly lower in DEV-ΔUL54 than in DPV-LoxP/DEV-ΔUL54 (Revertant) at 12 h, whereas these levels increased from 60–72 h. This finding implied that the pattern of UL54 regulation of the translation of viral genes is as follows: inhibition in the early stage and promotion in the middle and late stages.

Regulation of DEV UL54 on viral gene translation. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at 400 TCID50. After extracting RNC, RT-PCR was performed to assess the relative translational mRNA levels of UL19, UL30, UL48, gC, gD and gK at different time points. *p < 0.05; **p < 0.01.
DEV UL54 promotes the export of UL30 mRNA
We investigated the export of UL30 mRNA via FISH. The results indicated that UL30 mRNA was only located in the nucleus in DEV-ΔUL54, whereas it was located in both the nucleus and the cytoplasm in wild-type and Revertant cells (Fig. 7), meaning that UL54 could prompt UL30 mRNA export.

Effect of DEV UL54 on the export of UL30 mRNA. DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant) at 200 TCID50 and subjected to a FISH assay to visualize the localization of UL30 mRNA.
Discussion
The multi-functionality of HSV-1 ICP27 during infection has been well characterized5, 21, 35, but few reports have assessed its homologue DEV UL54. DEV UL54 is one of the immediate-early genes, which always encode proteins that are critical for regulation during infection. Therefore, we decided to study the regulatory role of DEV UL54 in viral gene expression.
Examination of the growth kinetics, plaque size and viral DNA copy number of three DEV-derived viruses generated by employing the Red recombination system showed a smaller plaque area, a lower viral titre and a lower viral DNA copy number for DEV-ΔUL54, which could be recovered. This finding indicated that the UL54 gene is important for DEV replication.
Next, the relative expression levels of UL19, UL30, UL48, gC, gD and gK, which belong to different genotypes and have different functions, were analysed by real-time PCR. The results showed that DEV UL54 could regulate viral gene expression either positively or negatively. To learn more about the effects of DEV UL54 on viral gene expression, the relative levels of mRNA transcription and translation were analysed after performing a nuclear run-off assay and RNC extraction, respectively. The results demonstrated that DEV UL54 could inhibit or augment viral gene transcription and translation. Interestingly, inhibition of UL30 by UL54 in the early stage did not cause a decrease in the total mRNA expression level, suggesting that UL54 may facilitate the export of UL30 mRNA, which was confirmed via a direct FISH assay. The shuttling property of the DEV UL54 protein may be important in this process.
The results for DEV UL54 in our study are consistent with reported findings for HSV-1 ICP276, 9, 10, 36, and high conservation may be responsible for this similarity. In the present study, we analysed gene expression only on the mRNA level; we did not study protein levels due to the lack of a polyclonal antibody. Although the analysis of translation may be a proxy to a certain extent, actual protein detection would be ideal. However, an “FRT” scar remained in the recombinant virus due to application of the Red recombination system, and the effects of an “FRT” scar on viral characteristics are unclear. A new system for the construction of recombinant viruses without a scar is thus being researched.
In summary, our results demonstrate that DEV UL54 could both positively and negatively regulate viral gene expression during transcription and translation and could promote the export of UL30 mRNA. Our research opens the door to studying the function of the UL54 gene, but our study was too shallow to characterize the detailed mechanisms of expression regulation by DEV UL54. Additionally, the domains responsible for regulation of DEV UL54 should also be examined.
Materials and Methods
Cells and viruses
Monolayer cultures of DEFs were grown in modified Eagle’s medium (MEM) supplemented with 10% new born bovine serum (NBS) and maintained at 37 °C in a 5% CO2 humidified incubator. Anti-UL54 polyclonal antibody, anti-UL13 polyclonal antibody, DEV-LoxP, DEV-ΔUL54, and DEV-ΔUL54 (Revertant) were all generated in our laboratory.
IFA and Western blotting
After incubation with DEFs for 2 h, any unadsorbed virus was discarded. The mixture of DEFs and DEV-derived virus was kept in MEM supplemented with 2% NBS and maintained at 37 °C in a 5% CO2 humidified incubator for 48 h.
To perform IFA, cells infected with DEV-ΔUL54 or DEV-ΔUL54 (Revertant) were sequentially treated with 4% paraformaldehyde, 0.5% Triton X-100, anti-UL54 polyclonal antibody and FITC-conjugated goat anti-rabbit IgG. Fluorescence microscopy was then applied to image the cells37.
Western blotting was performed as per standard protocols38. First, the cells infected with DEV-ΔUL54 or DEV-ΔUL54 (Revertant) were lysed, and the proteins were separated. Second, the separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes. After blocking with BSA and incubation with anti-UL54 polyclonal antibody and goat anti-rabbit HRP-labelled IgG, the membranes were developed using a DAB kit (TIANGEN, PA110).
Plaque formation assay
The plaque assay was also performed on DEFs, as per standard protocols39. After inoculation with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant), the DEFs were incubated on semisolid culture medium (a 2 × MEM and methylcellulose mixture) at 37 °C for several days. The cells were then fixed with paraformaldehyde and stained with 0.5% crystal violet. Plaque areas were measured with Image-Pro Plus 6.0 (IPP 6.0), and 100 plaques were randomly chosen for each virus.
Real-time fluorescence quantitative PCR (RT-PCR)
DEFs were infected with DEV-LoxP, DEV-ΔUL54 or DEV-ΔUL54 (Revertant). The cells were then harvested at 12, 24, 36, 48, 60, or 72 h for viral DNA replication and mRNA expression analyses. All analyses were performed independently in triplicate, and statistical significance was evaluated with the use of the unpaired Student’s t test.
To analyse DNA replication, a standard curve was first constructed. The viral DNA in the samples was then extracted according to the instructions of the Viral RNA/DNA Extraction Kit (Takara, 9766), and real-time PCR was performed with the purified nucleic acids as a template. All of the analyses were performed independently in triplicate, and statistical significance was evaluated with the use of the t test.
A nuclear run-off assay12, 40 and RNC extraction41,42,43 were performed on the samples before RNA isolation to investigate transcription and translation, respectively. For the nuclear run-off assay, nuclei were isolated according to the procedure of the nuclear extraction kit (Solarbio, SN0020). The infected cells were washed with PBS twice and centrifuged at 800 g for 5 min, and the cell pellet was harvested, after which the cells were resuspended in 1.0 mL of pre-cooled lysis buffer and pestled with a homogenizer for 10 sec after adding 50 μL of Reagent A. After centrifugation at 4 °C and 700 g for 5 min, the supernatant was discarded, and the sample was resuspended in 0.5 mL of pre-cooled lysis buffer. The suspension was then added to a centrifuge tube containing 0.5 mL of medium buffer and centrifuged at 4 °C and 700 g for 5 min. After the supernatant was discarded, 0.5 mL of lysis buffer was added, and the solution was centrifuged at 1000 g for 10 min. The supernatant was again discarded, and the pure nuclei were precipitated at the bottom of the tube. Finally, the nuclei were resuspended in 300 µL of runoff buffer (25 mM Tris-Cl (pH 8.0), 12.5 mM MgCl2, 750 mM KCl, and 1.25 mM NTP mix) and incubated at 37 °C for 15 min to complete transcription. The RNC was extracted using a common approach: infected cells were pretreated with 100 mg/mL CHX for 15 min and then washed with pre-chilled PBS. The cells were subsequently incubated in cell lysis buffer on ice for 30 min and centrifuged at 4 °C and 16200 r/min for 10 min to remove the cell debris. The supernatant was transferred to the surface of a sucrose buffer and centrifuged at 4 °C and 185000 r/min for 5 h to obtain the RNC. Total RNA, nuclear RNA, and RNC-containing RNA were isolated using RNAiso Plus (Takara, D9108A) and were reverse transcribed to cDNA (Takara, DRR047A), which served as a template for the subsequent real-time PCR. The primers used in this study are available upon request (Table 1). The relative transcription levels of the DEV UL19, UL30, UL48, gC, gD and gK genes were calculated using the 2−ΔCt method.
FISH assay
To conduct FISH, a probe was designed and synthetized by Sangon Biotech to visualize DEV UL30 mRNA. The sequence was 5′-TAGAGTCCCCAACAGATGCGAAAAGTAGTAGTCGGTG-3′, which was tagged with FITC at the 5′ terminus. DEF cells infected with DEV-derived virus were plated onto coverslips and sequentially treated with 4% paraformaldehyde, 0.2 mol/L HCl, and 100 μg/mL protease K. After prehybridization, hybridization and DAPI staining, the cells were imaged with a fluorescence microscope44.
References
Metwally, S. A. In Diseases of Poultry (eds David, E. Swayne et al.) 431–440 (John Wiley & Sons, Inc., 2013).
Wu, Y. et al. Complete genomic sequence of Chinese virulent duck enteritis virus. Journal of virology 86, 5965, doi:10.1128/JVI.00529-12 (2012).
Li, Y. et al. Molecular characterization of the genome of duck enteritis virus. Virology 391, 151–161 (2009).
Wang, J., Höper, D., Beer, M. & Osterrieder, N. Complete genome sequence of virulent duck enteritis virus (DEV) strain 2085 and comparison with genome sequences of virulent and attenuated DEV strains. Virus research 160, 316–325 (2011).
Smith, R. W., Malik, P. & Clements, J. B. The herpes simplex virus ICP27 protein: a multifunctional post-transcriptional regulator of gene expression. Biochemical Society transactions 33, 499–501, doi:10.1042/BST0330499 (2005).
Sacks, W. R., Greene, C. C., Aschman, D. P. & Schaffer, P. A. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J Virol 55, 796–805 (1985).
Sekulovich, R., Leary, K. & Sandri-Goldin, R. The herpes simplex virus type 1 alpha protein ICP27 can act as a trans-repressor or a trans-activator in combination with ICP4 and ICP0. Journal of virology 62, 4510–4522 (1988).
Hardwicke, M. A., Vaughan, P. J., Sekulovich, R. E., O’Conner, R. & Sandri-Goldin, R. M. The regions important for the activator and repressor functions of herpes simplex virus type 1 alpha protein ICP27 map to the C-terminal half of the molecule. Journal of virology 63, 4590–4602 (1989).
Rice, S. A., Su, L. S. & Knipe, D. M. Herpes simplex virus alpha protein ICP27 possesses separable positive and negative regulatory activities. Journal of virology 63, 3399–3407 (1989).
Su, L. & Knipe, D. M. Herpes simplex virus alpha protein ICP27 can inhibit or augment viral gene transactivation. Virology 170, 496–504 (1989).
Strain, A. K. & Rice, S. A. Phenotypic suppression of a herpes simplex virus 1 ICP27 mutation by enhanced transcription of the mutant gene. Journal of virology 85, 5685–5690, doi:10.1128/JVI.00315-11 (2011).
Jean, S., LeVan, K. M., Song, B., Levine, M. & Knipe, D. M. Herpes simplex virus 1 ICP27 is required for transcription of two viral late (gamma 2) genes in infected cells. Virology 283, 273–284, doi:10.1006/viro.2001.0902 (2001).
Dai-Ju, J. Q., Li, L., Johnson, L. A. & Sandri-Goldin, R. M. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. Journal of virology 80, 3567–3581, doi:10.1128/JVI.80.7.3567-3581.2006 (2006).
Li, L., Johnson, L. A., Dai-Ju, J. Q. & Sandri-Goldin, R. M. Hsc70 focus formation at the periphery of HSV-1 transcription sites requires ICP27. PloS one 3, e1491, doi:10.1371/journal.pone.0001491 (2008).
Kalamvoki, M. & Roizman, B. The histone acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. Journal of virology 85, 9472–9477, doi:10.1128/JVI.00876-11 (2011).
Sciabica, K. S., Dai, Q. J. & Sandri-Goldin, R. M. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. The EMBO journal 22, 1608–1619, doi:10.1093/emboj/cdg166 (2003).
Lindberg, A. & Kreivi, J. P. Splicing inhibition at the level of spliceosome assembly in the presence of herpes simplex virus protein ICP27. Virology 294, 189–198, doi:10.1006/viro.2001.1301 (2002).
Bryant, H. E., Wadd, S. E., Lamond, A. I., Silverstein, S. J. & Clements, J. B. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. Journal of virology 75, 4376–4385, doi:10.1128/JVI.75.9.4376-4385.2001 (2001).
McLauchlan, J., Phelan, A., Loney, C., Sandri-Goldin, R. M. & Clements, J. B. Herpes simplex virus IE63 acts at the posttranscriptional level to stimulate viral mRNA 3′ processing. Journal of virology 66, 6939–6945 (1992).
Nojima, T. et al. Herpesvirus protein ICP27 switches PML isoform by altering mRNA splicing. Nucleic acids research 37, 6515–6527 (2009).
Sandri-Goldin, R. M. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future microbiology 6, 1261–1277, doi:10.2217/fmb.11.119 (2011).
Cheung, P., Ellison, K. S., Verity, R. & Smiley, J. R. Herpes simplex virus ICP27 induces cytoplasmic accumulation of unspliced polyadenylated alpha-globin pre-mRNA in infected HeLa cells. Journal of virology 74, 2913–2919 (2000).
Ingram, A., Phelan, A., Dunlop, J. & Clements, J. B. Immediate early protein IE63 of herpes simplex virus type 1 binds RNA directly. The Journal of general virology 77(Pt 8), 1847–1851, doi:10.1099/0022-1317-77-8-1847 (1996).
Phelan, A., Dunlop, J. & Clements, J. B. Herpes simplex virus type 1 protein IE63 affects the nuclear export of virus intron-containing transcripts. Journal of virology 70, 5255–5265 (1996).
Sandri-Goldin, R. M. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes & development 12, 868–879 (1998).
Koffa, M. D. et al. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. The EMBO journal 20, 5769–5778, doi:10.1093/emboj/20.20.5769 (2001).
Chen, I.-H. B., Sciabica, K. S. & Sandri-Goldin, R. M. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. Journal of virology 76, 12877–12889 (2002).
Ellison, K. S., Maranchuk, R. A., Mottet, K. L. & Smiley, J. R. Control of VP16 translation by the herpes simplex virus type 1 immediate-early protein ICP27. Journal of virology 79, 4120–4131, doi:10.1128/JVI.79.7.4120-4131.2005 (2005).
Larralde, O. et al. Direct stimulation of translation by the multifunctional herpesvirus ICP27 protein. Journal of virology 80, 1588–1591, doi:10.1128/JVI.80.3.1588-1591.2006 (2006).
Fontaine-Rodriguez, E. C. & Knipe, D. M. Herpes simplex virus ICP27 increases translation of a subset of viral late mRNAs. Journal of virology 82, 3538–3545, doi:10.1128/JVI.02395-07 (2008).
Uprichard, S. L. & Knipe, D. M. Herpes simplex ICP27 mutant viruses exhibit reduced expression of specific DNA replication genes. Journal of virology 70, 1969–1980 (1996).
Liu, C., Cheng, A. & Wang, M. Bioinformatics Analysis of the Duck Enteritis Virus UL54 Gene. Research Journal of Applied Sciences Engineering & Technology 7, 2813–2817 (2014).
Liu, C. et al. Duck enteritis virus UL54 is an IE protein primarily located in the nucleus. Virology journal 12, 1 (2015).
Liu, C. et al. Characterization of nucleocytoplasmic shuttling and intracellular localization signals in Duck Enteritis Virus UL54. Biochimie (2016).
Sandri-Goldin, R. M. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Frontiers in bioscience: a journal and virtual library 13, 5241–5256 (2008).
McMahan, L. & Schaffer, P. A. The repressing and enhancing functions of the herpes simplex virus regulatory protein ICP27 map to C-terminal regions and are required to modulate viral gene expression very early in infection. Journal of virology 64, 3471–3485 (1990).
Zhang, S. et al. Characterization of duck enteritis virus UL53 gene and glycoprotein K. Virology journal 8, 1–9 (2011).
Luttmann, W., Kai, B., Küpper, M. & Myrtek, D. Western-Blot. (Springer Berlin Heidelberg, 2014).
Plaque Assay. (Springer Berlin Heidelberg, 2016).
Wang, Z. Nuclear Run-off Transcriptional Assay. Basic Medical Sciences & Clinics (1990).
Noriega, T. R. Signal Recognition Particle Interactions With Ribosome Nascent Chain Complexes. Dissertations & Theses - Gradworks (2014).
Zhong, J. et al. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. Plos Genetics 11 (2015).
Zhong, J. et al. Resolving chromosome-centric human proteome with translating mRNA analysis: a strategic demonstration. Journal of Proteome Research 13, 50–59 (2014).
Bayani, J. & Squire, J. A. Fluorescence in situ Hybridization (FISH). (Springer, 2009).
Acknowledgements
This research was financially supported with grants from the National Science and Technology Support Program for Agriculture (2015BAD12B05), the China Agricultural Research System (CARS-43–8), Special Fund for Key Laboratory of Animal Disease and Human Health of Sichuan Province (2016JPT0004).
Author information
Authors and Affiliations
Contributions
C.L. conceived, designed and performed the experiments, analyzed the data and wrote the paper; A.C., M.W. conceived and designed the experiments; S.C., R.J., D.Z., M.L., K.S., Q.Y., Y.W., X.Z., X.C., interpreted the data. All authors read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, C., Cheng, A., Wang, M. et al. Regulation of viral gene expression by duck enteritis virus UL54. Sci Rep 7, 1076 (2017). https://doi.org/10.1038/s41598-017-01161-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01161-0
This article is cited by
-
Duck plague virus UL41 protein inhibits RIG-I/MDA5-mediated duck IFN-β production via mRNA degradation activity
Veterinary Research (2022)
-
The Pivotal Roles of US3 Protein in Cell-to-Cell Spread and Virion Nuclear Egress of Duck Plague Virus
Scientific Reports (2020)
-
Molecular characterization of duck enteritis virus UL41 protein
Virology Journal (2018)
-
Duck plague virus Glycoprotein J is functional but slightly impaired in viral replication and cell-to-cell spread
Scientific Reports (2018)
-
US10 Protein Is Crucial but not Indispensable for Duck Enteritis Virus Infection in Vitro
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
