
Abstract
Staphylococcus aureus is the leading cause of skin and soft tissue infections (SSTIs) and mounting antibiotic resistance requires innovative treatment strategies. S. aureus uses secreted cyclic autoinducing peptides (AIPs) and the accessory gene regulator (agr) operon to coordinate expression of virulence factors required for invasive infection. Of the four agr alleles (agr types I-IV and corresponding AIPs1-4), agr type I isolates are most frequently associated with invasive infection. Cyclization via a thiolactone bond is essential for AIP function; therefore, recognition of the cyclic form of AIP1 may be necessary for antibody-mediated neutralization. However, the small sizes of AIPs and labile thiolactone bond have hindered vaccine development. To overcome this, we used a virus-like particle (VLP) vaccine platform (PP7) for conformationally-restricted presentation of a modified AIP1 amino acid sequence (AIP1S). Vaccination with PP7-AIP1S elicited AIP1-specific antibodies and limited agr-activation in vivo. Importantly, in a murine SSTI challenge model with a highly virulent agr type I S. aureus isolate, PP7-AIP1S vaccination reduced pathogenesis and increased bacterial clearance compared to controls, demonstrating vaccine efficacy. Given the contribution of MRSA agr type I isolates to human disease, vaccine targeting of AIP1-regulated virulence could have a major clinical impact in the fight against antibiotic resistance.
Similar content being viewed by others

Introduction
Between 2000 and 2012, the incidence of skin and soft tissue infections (SSTI) in the USA is estimated to have increased 40%, with treatment expenditures increasing from $4.4 billion to $13.8 billion in 2012 dollars1. Among emergency room patients, the majority of SSTIs are caused by Staphylococcus aureus, and over half of these isolates are methicillin-resistant (MRSA)2, 3. Compared to antibiotic-susceptible strains, MRSA SSTI treatment failure requires added interventions with associated increases in human suffering and medical costs4. Given the ongoing antibiotic resistance crisis, the recurrent nature of S. aureus SSTI5, and the lack of an approved S. aureus vaccine to date6, there is an urgent need for alternative approaches to combat infections caused by MRSA.
The production of virulence factors required for S. aureus SSTI is largely regulated by the accessory gene regulator operon (agr)7, 8 through a bacterial communication system known as quorum sensing. Induction of agr signaling depends upon the accumulation of small, secreted autoinducing peptides (AIPs) to activate a receptor histidine kinase, AgrC, in the bacterial cell membrane9, 10. AgrC activation drives downstream production of the effector molecule, RNAIII, which in turn regulates expression of over 200 virulence genes contributing to invasive infection7. S. aureus isolates express one of four agr alleles (agr-I to agr-IV), with each secreting a unique AIP (AIP1-AIP4) and expressing a corresponding AgrC. Previously, both an anti-AIP4 monoclonal antibody (mAb)11, 12 and an AIP4 immunologic mimotope vaccine13 showed protection against infection caused by agr type IV isolates. However, antibody or vaccine targeting of signaling by agr type I isolates, which are most associated with invasive S. aureus infection14, 15, has not been reported.
S. aureus AIP1 is an eight amino acid peptide (YSTCDFIM) cyclized by a thiolactone bond between the Cys4 side-chain and the carboxyl group of the C-terminal residue (Met8) (Fig. 1a). Given that cyclization is essential for function, immune recognition of the cyclic form of AIP1 may be necessary for antibody-mediated neutralization. However, the small size of these peptides makes them innately non-immunogenic and, together with the labile nature of the thiolactone, increases the difficulty of vaccine development12, 13, 16. We sought to overcome these challenges using a bacteriophage virus-like particle (VLP) vaccine platform. These VLPs self-assemble from recombinantly expressed bacteriophage coat proteins which can be genetically altered for surface presentation of practically any epitope in a multivalent format that virtually guarantees strong immunogenicity resulting in high titer, high affinity, and long-lasting antibodies17. Specifically, we hypothesized that a vaccine produced by conformationally-restricted presentation of the AIP1 amino acid sequence on the surface of bacteriophage VLPs would elicit antibodies against native AIP1 and induce immune control of agr type I-regulated virulence.

Design and preparation of PP7-AIP1S VLPs. (a) Schematic of AIP1 and amino acid sequence of AIP1-C4S (AIP1S). (b) Ribbon representation of the PP7 coat protein dimer (one monomer is shown in green and the other in magenta) which can be expressed as a single-chain dimer. Depicted is the first AB loop (indicated by arrow) and the AIP1S sequence (spheres) modeled into the second AB loop (PDB ID 2QUD21) using GalaxyWeb29, 30. Image prepared using PyMol (PyMOL molecular graphics system, version 1.5.0.4; Schrodinger, LLC). (c) Schematic of the site of AIP1S insertion into the second AB loop of the PP7 single chain dimer. (d) Coomassie-stained 16% SDS-PAGE showing the relative size of the PP7 single-chain dimer compared to PP7 with the AIP1S insert.
To test this, we produced a VLP-based agr type I vaccine by cloning a modified AIP1 amino acid sequence (YSTSDFIM) into an immuno-prominent surface loop (the AB-loop) of the Pseudomonas aeruginosa RNA bacteriophage PP7 coat protein18,19,20,21. As expected, the resulting vaccine (PP7-AIP1S) elicited antibodies which recognized AIP1 in vitro and was efficacious in a murine SSTI model upon challenge with a highly virulent MRSA agr type I isolate. Compared to controls, PP7-AIP1S vaccination resulted in reduced agr function and agr-regulated virulence factor production at the site of infection. Importantly, PP7-AIP1S vaccination significantly reduced S. aureus pathogenesis, based on dermonecrosis and weight loss, and increased bacterial clearance, findings consistent with enhanced host innate defense in the absence of agr function8, 22,23,24,25,26. Together, these results demonstrate the protective benefits of vaccine-induced immune control of agr type I-regulated virulence. Given that several important pathogens utilize similar structurally constrained peptides for virulence regulation27, our findings highlight the potential clinical utility of VLP-based vaccines targeting virulence regulators as an alternative or adjunct approach to combat infections caused by other human pathogens.
Results
Presentation of the S. aureus AIP1 sequence on VLPs induces AIP1-recognizing antibodies
We previously found that vaccination with AIP1 (cyclic or linear) chemically cross-linked to VLPs did not protect mice against subsequent skin infection (unpublished data). This could have been at least in part due to instability of the AIP thiolactone bond12 resulting in peptide linearization at some time during the vaccine preparation or vaccination process13. Therefore, in an effort to promote immunogenicity and maintain the structural integrity of AIP1 presentation to the adaptive immune system, we inserted a modified AIP1 sequence to be presented in a highly-constrained β-turn on the surface of VLPs assembled from the previously reported PP7 single-chain coat protein dimer18,19,20, 28,29,30 (Fig. 1a–c). The icosahedral capsid of the Pseudomonas aeruginosa RNA bacteriophage PP7 self-assembles from 180 coat protein monomers31, 32, whose structural arrangement is equivalent to 90 coat protein dimers31. Recombinantly-expressed PP7 coat protein self-assembles into stable VLPs consistent in size with the bacteriophage PP7 icosahedral capsid28, 31, 33. The highly constrained β-turn within the PP7 coat protein, called the AB-loop, is displayed on the surface of the assembled capsid21, 31, 32 or VLP, and peptides inserted into this loop are highly immunogenic20, 28, 33, 34. To avoid potential intermolecular disulfide bond formation via the AIP1 internal cysteine that could negatively impact VLP purification and immune presentation, the inserted AIP1 sequence included a cysteine to serine mutation in position 4 (YSTCDFIM to YSTSDFIM) (referred to as AIP1S) (Fig. 1a). We predicted that this conservative mutation would still allow vaccine induction of AIP1 specific antibodies given (i) that the cysteine side chain is confined to the thiolactone bond in the native molecule, and (ii) that presentation of the AIP1 sequence in the context of the highly constrained PP7 AB-loop would simulate the cyclic nature of AIP1 in the absence of this labile bond. Recombinantly-expressed PP7-AIP1S VLPs resolved into a single peak by size exclusion purification, with agarose gel electrophoresis showing fractions containing a single protein band co-localizing with encapsidated RNA, and with homogeneity shown by dynamic light scattering analysis (Supplemental Fig. S1). Insertion of the AIP1S sequence into the PP7 single-chain coat protein dimer was verified by DNA sequencing of the expression plasmid (data not shown) and by electrophoretic size comparison (Fig. 1d). Given that the equivalent of 90 coat protein dimers are needed to form the PP7 bacteriophage icosahedral capsid, PP7-AIP1S VLPs produced from PP7 single-chain coat protein dimers with the AIP1S insertion in the second AB-loop should therefore display 90 copies of AIP1S for immune stimulation.
We first sought to determine whether vaccination with PP7-AIP1S would induce production of antibodies capable of recognizing S. aureus AIP1. To address this, we vaccinated mice with PP7-AIP1S (twice with a 4-week interlude) and then measured the ability of serum antibodies to recognize AIP1. Although prior vaccination efforts using linear or cyclic AIP1 chemically cross-linked to VLPs induced antisera capable of binding immobilized linear or cyclic AIP1 by ELISA (unpublished data), these vaccines failed to provide protection during S. aureus infection13, possibly suggesting that antibody recognition of the AIP linear tail (Fig. 1a) or linearized AIP is not sufficient for protection. Therefore, here we measured antibody binding to AIP1S as presented in the AB-loop on the surface of PP7, followed by competition binding to soluble, cyclic AIP1. Serum collected at two-, four- and eight-weeks after the last vaccination with PP7-AIP1S, but not after PP7 control vaccination, showed dose-dependent binding to the AIP1S sequence present on PP7-AIP1S VLPs (Fig. 2a and Supplemental Fig. S2). Importantly, in competitive dose-response assays, AIP1S binding by eight-week post-vaccination antiserum (geometric mean titer = 4,550) was inhibited by synthetic cyclic AIP1, but not synthetic AIP2 (GVNACSSLF) (Fig. 2b), demonstrating specificity and the ability to bind native AIP1. Therefore, these results demonstrate that presentation of AIP1S, which lacks the native AIP thiolactone bond, within the PP7 AB-loop is sufficient to elicit antibodies which recognize soluble, native AIP1.

PP7-AIP1S vaccination induces antibodies which recognize soluble AIP1. BALB/c mice were vaccinated twice (i.m.) at 4 week intervals with 10 µg of PP7-AIP1S or PP7 wild-type (control). (a) Serum was collected at the indicated time points after the second vaccination. Serum was then pooled (n = 3 mice per group), treated as described in Materials and Methods, and relative binding to PP7-AIP1S determined by ELISA. (b) PP7-AIP1S antiserum collected at eight weeks after the second vaccination was prepared as in (a), and relative AIP1S binding determined in the presence and absence of the indicated concentrations of AIP1 or AIP2 (n = 3 mice per group; duplicate experiments performed in triplicate). Data are mean ± s.e.m. Kruskal-Wallis ANOVA p < 0.0001 with Dunn’s post-test: *p < 0.05; ***p < 0.001.
PP7-AIP1S vaccination provides protection in a murine model of S. aureus dermonecrosis
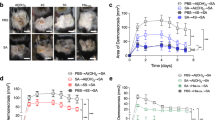
MRSA isolates of the pulsed-field gel electrophoresis type USA300 (agr type I) have long been the cause of most community-associated MRSA (CA-MRSA) infections, and now also cause an increasing number of health-care associated infections35. In mouse models of USA300 SSTI, infection with an isogenic agr-deletion mutant (Δagr) results in significantly decreased pathogenesis and increased bacterial clearance compared to infection with the wild-type agr+ strain8, 22,23,24,25. Therefore, we postulated that vaccination with PP7-AIP1S would induce immune suppression of agr-signaling in vivo, thus reducing pathogenesis and increasing bacterial clearance during SSTI. To evaluate the efficacy of PP7-AIP1S vaccination against agr type I-mediated virulence and to avoid potential non-specific effects of VLP administration36, we challenged mice eight weeks after final vaccination using a well-established mouse model of S. aureus SSTI37 and the highly virulent USA300 isolate LAC38. As expected, PP7-AIP1S vaccinated mice showed reduced abscess formation and dermonecrosis over the course of a six-day infection compared to controls (Fig. 3a–d), although differences in weight loss (used as a measure of morbidity) did not reach statistical significance (Fig. 3e). Importantly, bacterial burden on day 6 post-infection was also significantly reduced in the PP7-AIP1S vaccinated group compared to both PBS and PP7 controls (Fig. 3f). Reduced bacterial burden was consistent with significantly lower local levels of the inflammatory cytokines IL-1β, TNFα, IL-1α and IL-17 in PP7-AIP1S vaccinated mice compared to PBS controls (Fig. 3g). However, differences in local levels of these cytokines between the PP7 WT and PP7-AIP1S groups did not reach significance. In addition, local IL-6 production was significantly reduced in both PP7 WT and PP7-AIP1S vaccinated mice, potentially pointing to a non-specific effect of VLP vaccination on pro-inflammatory cytokine production at this time point. In contrast, significant differences were not observed in local production of CXCL1 or the anti-inflammatory cytokine IL-10. Together, these data demonstrate the efficacy of PP7-AIP1S vaccination against S. aureus agr type I-regulated pathogenesis during skin infection.

PP7-AIP1S vaccination limits the severity of S. aureus skin infection in a mouse model of dermonecrosis. BALB/c mice were vaccinated twice (i.m.) at 4 week intervals with 10 µg of the indicated VLPs or PBS control. Eight weeks after the second vaccination, mice were challenged by subcutaneous infection with 4 × 107 CFU of USA300 LAC. Representative (a) day 3 images of infection site and (b) daily measures of abscess area and dermonecrosis. Calculated area under the curve (AUC) values for (c) abscess area (ANOVA p < 0.0042), (d) dermonecrosis (p = 0.0177) and (e) percent weight change over the six day infection, as well as (f) day 6 bacterial burden at the site of infection (p = 0.0001) (representative of two independent experiments of n = 6 mice per group). (g) Cytokine levels in clarified abscess tissue homogenate on day 6 post-infection (ANOVA IL-1β, p = 0.0587; TNFα, p = 0.0358; IL-1α, p = 0.0171; IL-17, p = 0.0322; IL-6, p = 0.0010) (n = 6 mice per group). Data are mean ± s.e.m. Newman-Keuls post test: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
PP7-AIP1S vaccination inhibits S. aureus agr-signaling in vivo
S. aureus agr-signaling induces expression of the effector molecule RNAIII as well as production of alpha-hemolysin (Hla), the causative agent of dermonecrosis39,40,41,42,43. The results of our challenge studies, as well as our in vitro studies showing that antibodies from PP7-AIP1S vaccinated mice bind soluble AIP1, suggested that vaccination with PP7-AIP1S results in immune suppression of agr-signaling during S. aureus SSTI. If correct, we would expect reduced RNAIII transcription and Hla expression at the site of infection (local) in PP7-AIP1S vaccinated mice compared to controls. To test this, we measured local RNAIII expression and Hla protein levels on days one and six, respectively, following subcutaneous infection. As expected, RNAIII expression was reduced at the site of infection in PP7-AIP1S vaccinated mice compared to controls (Fig. 4a). Furthermore, day six post-infection Hla levels were reduced in PP7-AIP1S vaccinated mice relative to PBS controls (Fig. 4b,c), although differences between PP7-AIP1S versus PP7 WT vaccination were not statistically significant. Together, these data support a mechanism of action whereby vaccination with PP7-AIP1S induces immune control of S. aureus agr type I signaling and virulence regulation during SSTI.

PP7-AIP1S vaccination limits agr function at the site of S. aureus infection. BALB/c mice were vaccinated twice (i.m.) at 4 week intervals with 10 µg of the indicated VLPs or PBS control. Eight weeks after the second vaccination, mice were challenged by subcutaneous infection with 4 × 107 CFU of USA300 LAC. (a) Local RNAIII transcription on day 1 post-infection measured by qPCR (n = 4 mice per group, Kruskal-Wallis ANOVA p = 0.0029). (b) Representative immunoblot (showing recombinant Hla, MW marker and 3 mice per group) and (c) quantification of Hla levels (relative to PBS control) in clarified abscess tissue homogenate on day 6 post-infection (n = 6 mice per group) (Kruskal-Wallis ANOVA p = 0.0025) with Dunn’s post-test: ns, not significant; *p < 0.05; **p < 0.01.
Discussion
The ongoing antibiotic resistance crisis highlights the urgent need for non-conventional approaches to combat infectious disease, including approaches to inhibit bacterial virulence44, 45. In the case of the important human pathogen Staphylococcus aureus, virulence regulation is largely mediated by the agr operon via secretion of AIPs9, 10. These small, conformationally-restrained, secreted peptides bind in an autocrine and paracrine fashion to the bacterial membrane receptor AgrC, which in turn regulates downstream virulence factor expression. Therefore, antibody-mediated sequestration of secreted AIPs could neutralize agr-signaling and virulence factor expression on a population level. Of the four S. aureus agr types, agr type I isolates are most frequently associated with invasive infection14, 15. Here we report that multivalent, conformationally-restricted presentation of a modified AIP1 amino acid sequence on VLPs elicits immune control of S. aureus agr type I-regulated virulence. Specifically, PP7-AIP1S vaccination (1) induced the production of anti-AIP1 antibodies, (2) limited agr type I-signaling in vivo and (3) demonstrated efficacy (reduced pathogenesis and increased bacterial clearance) in a mouse model of S. aureus SSTI. Given these results and the contribution of agr type I isolates to human S. aureus infection14, 15, vaccine prevention of agr type I-mediated virulence could have a major clinical impact and make a significant contribution to the fight against antibiotic resistance.
The diversity of virulence factors produced by S. aureus 7, many of which disable innate immune cells46,47,48, and the range of infection types (skin, pneumonia, bacteremia, etc.)7, 49, suggests that multiple anti-virulence approaches may be needed to limit human disease. For example, targeting specific virulence factors, in particular Hla which is a major contributor to pathogenesis50, has shown efficacy in numerous animal models40, 41, 51,52,53,54 and a monoclonal antibody targeting Hla (MEDI4893) is currently in human clinical trials55. Broader approaches aimed at inhibiting S. aureus virulence regulation have included peptide and small molecule targeting of the agr system22, 24, 26, 56,57,58,59,60,61,62,63,64,65,66,67,68, as well as development of a monoclonal antibody (mAb) against S. aureus AIP412, 69. However, agr-signaling has been shown to occur early post-infection and disruption of this early signaling correlates with reduced pathogenesis in the host22, suggesting a possible limit to the window of opportunity for therapeutic agr-inhibition. Therefore, the development of an efficacious anti-agr vaccine could expand the impact of S. aureus virulence regulation strategies to have the broadest potential clinical benefit to patients. In this regard, we previously developed a VLP-based AIP4 mimotope vaccine by screening a VLP-peptide library against an anti-AIP4 mAb, AP4-24H1112, 13, 69, shown by passive transfer to be protective in a mouse model of agr type IV SSTI. Here we advance this work by demonstrating the efficacy of PP7-AIP1S vaccination against S. aureus agr type I-regulated virulence using the highly virulent CA-MRSA USA300 isolate LAC38. While it will be important in future work to expand these studies to include infections using other agr type I isolates versus isolates of heterologous agr types, our in vivo results, together with in vitro data demonstrating that PP7-AIP1S elicited antibodies bind soluble AIP1, but not soluble AIP2, suggest that protection afforded by PP7-AIP1S vaccination is specific to agr type-I regulated virulence. Therefore, this VLP-based approach may be utilized to produce a combined vaccine against virulence regulation by each of the agr types, thus serving as a valuable component of an overall anti-virulence strategy.
In addition to Staphylococcal species9, 10, other human pathogens using agr-like quorum sensing systems and secreted peptides to coordinate virulence factor expression27 could be targeted by VLP-based vaccination. For example, the food-borne pathogen Listeria monocytogenes uses a variety of communication systems to regulate virulence70, 71, including an agr locus and recently identified secreted AIP72,73,74,75,76. In L. monocytogenes, the agr system regulates over 650 genes contributing to virulence including ones involved in biofilm formation and host cell invasion73. Similarly, Enterococcus faecalis, an important cause of drug resistant infections77, uses the agr-like fsr gene locus and the secreted, cyclic peptide gelatinase biosynthesis-activating pheromone (GBAP)78, 79 to regulate expression of virulence factors important for biofilm formation and pathogenesis80,81,82,83,84,85. Importantly, it has also recently been shown that an agr locus regulates production of toxins A and B by the multidrug resistant pathogen Clostridium difficile 86, 87. These C. difficile toxins are directly responsible for disease manifestation88 which, in severe cases, can result in sepsis and death89, suggesting that interference with agr-signaling by this pathogen could significantly limit disease. Therefore, a VLP-vaccine platform could provide a straight-forward approach to elicit immune inhibition of agr- and agr-like virulence signaling by these and other important human pathogens.
While vaccine targeting of S. aureus agr-regulated virulence has clear benefits, potential limitations have also been noted11, 90, 91. For example, in vitro studies have shown an isolate-dependent association between deletion of agr and increased biofilm formation, raising the possibility that inhibiting agr in vivo could promote biofilms92,93,94. Increased in vitro biofilm formation by agr deletion mutants is attributed to increased expression of genes encoding surface associated factors, such as protein A and the fibronectin binding proteins (FnBPs), in parallel with down regulation of secreted virulence factors which promote biofilm dispersal7, 24, 95,96,97,98. However, agr-regulated factors can inhibit phagocyte clearance of S. aureus in biofilms99, suggesting that targeting agr in vivo may also support host innate defense against biofilm infections. A second concern regarding in vivo inhibition of agr is whether this will select for agr mutants. Although most clinical isolates of S. aureus are agr positive, isolates with mutations in agr can arise naturally during chronic infections15, 100, 101. However, whether antibody-mediated inhibition of AIP is more likely to select for agr mutants than host innate effectors which antagonize agr-signaling, such as apolipoprotein B (apoB)23, 25, hemoglobin102 and reactive oxygen species103, has yet to be addressed. Given the contribution of agr to S. aureus pathogenesis and the protection afforded by vaccine targeting of AIPs, the potential in vivo impact of this approach on both S. aureus biofilm formation and the selection of agr mutants are important areas for future investigation.
Virus-like particles have proven to be a flexible and highly immunogenic platform for vaccine design, and are currently used in FDA-approved vaccines104, including Hepatitis B vaccines105 and the current nonavalent HPV vaccine (Gardasil 9) designed to induce protection against nine HPV types106. Although non-replicating, the dense, repetitive array of coat proteins comprising VLPs is largely unique to microbial antigens and this multivalency triggers a robust immune response in mammals. Therefore, VLPs can dramatically increase the immunogenicity of otherwise poorly immunogenic peptides17, 107 including self-antigens108, 109. This property, along with the potential for presentation of conformation-dependent antigens, has resulted in investigation of VLP-based vaccines against numerous pathogenic viruses, allergies, cancer, autoimmune disease, Alzheimer’s disease and chronic diseases such as hypertension17, 110,111,112,113. However, reports of the use of VLP-based vaccines to elicit adaptive immunity against specific bacterial pathogens or proteins have come mainly from our own work13 and from research targeting Streptococcal species114,115,116, suggesting that the flexibility of VLP-based vaccine approaches to address bacterial diseases remains largely untapped. Given the FDA approval and success of VLP vaccines against viral pathogens, the use of VLP-based vaccines to prevent infections by the many important human bacterial pathogens warrants further investigation.
In this era of diminishing antibiotic efficacy, a multi-pronged approach, including novel antibiotics, host-targeted therapeutics, vaccines, anti-virulence strategies and combined therapies will likely be crucial for combating disease caused by antibiotic resistant pathogens11, 44, 45, 117. Here we present a novel approach to achieve vaccine induced immune control of S. aureus agr-regulated virulence. This work highlights the potential clinical utility of VLP-based vaccines as part of an overall strategy to combat infections caused by MRSA and other important antibiotic resistant human pathogens utilizing secreted peptides for virulence regulation27.
Methods
Ethics statement
Animal studies described herein were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of New Mexico Health Sciences Center (Animal Welfare Assurance number D16-00228) and conducted in strict accordance to recommendations in the Guide for the Care and Use of Laboratory Animals 118, the Animal Welfare Act, and U.S. federal law.
Bacterial strains and growth conditions
The CA-MRSA USA300 isolate LAC38 (generously provided by Dr. Frank DeLeo, Rocky Mountain National Laboratories, National Institutes of Health, Hamilton, MT) was used for infection studies. Early exponential-phase bacteria were prepared as previously described119 and stored at −80 °C for no more than two weeks prior to use. For infection studies, bacteria were diluted in USP-grade saline (B. Braun Medical, Irvine, CA) to yield 4 × 107 CFU per 50 µL. The number of CFU was verified by plating ten-fold serial dilutions onto Trypticase soy agar containing 5% sheep blood (Becton, Dickinson and Company; Franklin Lakes, NJ).
VLP cloning, expression and purification
The pDSP7K plasmid120, encoding the PP7 single-chain dimer under the T7 promoter and transcription terminator, was used for synthesis of PP7-AIP1S VLPs in E. coli. With pDSP7K as a template, PCR was used to produce an insert fragment encoding a KpnI restriction site, the modified AIP1 sequence (YSTSDFIM), and a downstream BamHI site (forward primer 5′-GGC GGT ACC TAC AGT ACC TCT GAC TTC ATC ATG GAG GCT ACT CGC ACT CTG ACT GAG-3′; reverse primer 5′-CGG GCT TTG TTA GCA GCC GG-3′). The PCR fragment was inserted into the pDSP7K at the KpnI and BamHI restriction sites and insertion was verified by sequence analysis.
E. coli C41 cells (Lucigen, Middleton, WI) transformed with pDSP7K or the pDSP7K-AIP1S expression plasmids were grown at 37 °C to an OD600 of 0.8. Expression was induced with 1 mM IPTG, cells cultured for an additional 3 hours, and harvested by centrifugation. Cell pellets were lysed and VLPs purified essentially as described previously20 but with size exclusion purification using a 16/60 Sephacryl S-400 HR column (GE Healthcare, Pittsburgh, PA). VLP purity was verified by SDS-PAGE and agarose gel electrophoresis plus Coomassie and ethidium bromide staining. VLPs were concentrated using Amicon Ultra Centrifugal filter units (100 K MWCO) (EMD Millipore, Billerica, MA), and concentrations determined by SDS-PAGE comparison to hen egg lysozyme concentration standards (Sigma-Aldrich, St. Louis, MO). Homogeneity was based on modality analysis using a Malvern Zetasizer Nano Z (Malvern, UK). VLP aliquots were stored at −20 °C until use.
Mouse immunizations
Four week old, female BALB/cJ mice (Jackson Laboratories, Bar Harbor, ME, USA) were immunized by injection into the caudal thigh muscle with 50 µL of PBS alone or containing 10 µg of either PP7-AIP1S or PP7. Mice received an identical injection four weeks after the initial dose. Serum for ELISA analysis was collected by cardiac puncture at two, four or eight weeks after the second vaccination, with challenge experiments performed at the eight week time-point.
ELISA
ELISA plates to measure serum antibody binding to AIP1S were prepared by coating Ultra Cruz ELISA High Binding plates (Santa Cruz Biotechnology, Santa Cruz, CA) with 125 ng per well of recombinant PP7 or PP7-AIP1S in 50 µL PBS and incubating 20 hours at room temperature (RT) with shaking. After removing excess liquid, plates were blocked for 2 hours with PBS containing 0.05% Tween-20 and 1% casein. To reduce PP7- and potential E. coli-binding antibodies (depleted serum) mouse serum was treated as follows: Serum was diluted 1:50 in PBS and incubated for one hour at RT with end-over-end rotation together with recombinant PP7 (10 µg per 300 µL diluted serum) and PBS-washed C41 cells (the E. coli strain used for VLP-expression) (~9 × 106 CFUs). The mixture was centrifuged (5 min at 11,600 × g) to remove antibody bound to C41 cells, and the intermediate depleted serum processed through an Amicon Ultra Centrifugal filter unit (100K MWCO) to remove antibody bound to PP7. The presence of antibody in the filtrate (depleted serum) was verified by SDS-PAGE and Coomassie staining. The final depleted serum was serially diluted onto PP7- or PP7-AIP1S-coated ELISA plates and incubated for 1 hour at RT. Murine antibodies bound to VLPs were detected using goat anti-mouse poly-HRP secondary antibody (ThermoFisher Scientific, Waltham, MA) and developed using 1-Step™ Ultra TMB-ELISA according to manufacturer’s directions (ThermoFisher Scientific). For each serum sample and dilution, AIP1S specific binding (ΔA450) was equal to the A450 for PP7-AIP1S binding minus the A450 for PP7 binding. For competition ELISAs, depleted serum was incubated for 1 hour at 37 °C with the indicated concentrations of AIP1 or AIP2 (BioPeptide Co., Inc., San Diego, CA) before addition to PP7-AIP1S-coated ELISA plates.
Mouse skin infection model
The mouse model of dermonecrosis was implemented essentially as previously described37. One to three days before infection (eight weeks after the second vaccination), Nair™ was used to depilate the right flank of the mice (site of infection). On the day of infection, mice were anesthetized by isoflurane inhalation and infected by subcutaneous injection of 50 µL of saline containing 4 × 107 CFU of LAC. Mice were weighed the day of injection and daily thereafter until sacrifice. Injection sites were photographed daily and abscess and dermonecrosis areas determined by analysis with ImageJ121. Six days after infection, mice were sacrificed by CO2 asphyxiation and a 2.25-cm2 section of skin surrounding the abscess was excised for mechanical disruption. Abscess homogenate was serially diluted and plated on sheep blood agar to determine infection site bacterial burden. The remaining homogenate was clarified by centrifugation and the clarified fraction stored at −80 °C until cytokine analysis.
Cytokine analysis by multiplex assay
Clarified abscess tissue homogenates were quick thawed at 37 °C and concentrations of the indicated cytokines determined using a BioPlex 200 system and BioPlex manager software (Bio-Rad, Hercules, CA) together with a custom-designed mouse multiplex assay (EMD Millipore, Billerica, MA) according to manufacturer’s directions.
RNA isolation from tissue and quantitative PCR analysis
For analysis of day one post-infection bacterial gene transcription, 2.25 -cm2 sections of skin surrounding the infection site were harvested, minced, and stored in RNAlater (Qiagen, Valencia, CA) at −20 °C. RNA was isolated using QIAzol (Qiagen) and purified using RNeasy kits (Qiagen) according to manufacturer’s directions. cDNA conversion from RNA was performed with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and specific primers for S. aureus 16S (reverse, 5′-TTC GCT CGA CTT GCA TGT A-3′) or RNAIII (reverse, 5′-GATGTTGTTTACGATAGCTTACATGC-3′) (Integrated DNA Technologies, Coralville, IA). Quantitative PCR (qPCR) was performed using a ViiA-7 RT-PCR system (Applied Biosystems), the specific primers and probes23 for 16S (forward primer, 5′-TGA TCC TGG CTC AGG ATG A-3′; reverse primer above and probe 5′-CGC TGG CGG CGT GCC TA-3′) and RNAIII (forward primer, 5′-AAT TAG CAA GTG AGT AAC ATT TGC TAG T-3′; reverse primer above and probe 5′-AGT TAG TTT CCT TGG ACT CAG TGC TAT GTA TTT TTC TT-3′) (Integrated DNA Technologies) and TaqMan Gene Expression Master Mix according to the manufacturer’s protocol (Applied Biosystems). Data are shown as the fold expression of RNAIII versus 16S and relative to the PBS control.
Tissue Hla quantification by Western blot
For Western blot analysis of Hla levels in clarified abscess homogenate, frozen samples were quick thawed and equal amounts of total protein (based on A280) were electrophoresed on 16% Tris-glycine SDS-PAGE gels (Life Technologies, Grand Island, NY). Following transfer to polyvinylidene fluoride membrane, membranes were blocked overnight at 4 °C with TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween 20) with 5% nonfat dry milk. Hla was detected using sheep anti-Hla primary antibody (ab15948, Abcam, Cambridge, MA) and alkaline phosphatase-conjugated rabbit polyclonal anti-sheep secondary. Membranes were developed with nitroblue tetrazolium (NBT)/5-bromo-4-chloro-3-indolyl-phosphate (BCIP) (Thermo Scientific). Band intensity relative to recombinant Hla control was measured on a FluorChem R system using AlphaView software (ProteinSimple, San Jose, CA).
Statistical analysis
GraphPad Prism version 5.04 (GraphPad Software, San Diego California) was used for all statistical evaluations. One-way ANOVA parameters followed Bartlett’s test for equal variances and were used with Bonferroni’s (ANOVA) or Dunn’s (Kruskal-Wallis test, non-parametrics) post-hoc multiple comparison analyses. Results were considered statistically significant at p < 0.05.
References
Lee, G. C. et al. Incidence and Cost of Skin and soft Tissue Infections In the united States. Value Health 18, doi:10.1016/j.jval.2015.03.1424 (2015).
Moran, G. J. et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355, 666–674, doi:10.1056/NEJMoa055356 (2006).
Talan, D. A. et al. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin. Infect. Dis. 53, 144–149, doi:10.1093/cid/cir308 (2011).
Labreche, M. J. et al. Treatment failure and costs in patients with methicillin-resistant Staphylococcus aureus (MRSA) skin and soft tissue infections: a South Texas Ambulatory Research Network (STARNet) study. J. Am. Board Fam. Med. 26, 508–517, doi:10.3122/jabfm.2013.05.120247 (2013).
Montgomery, C. P., David, M. Z. & Daum, R. S. Host factors that contribute to recurrent staphylococcal skin infection. Curr. Opin. Infect. Dis. 28, 253–258, doi:10.1097/QCO.0000000000000156 (2015).
Fowler, V. G. Jr. & Proctor, R. A. Where does a Staphylococcus aureus vaccine stand? Clin. Microbiol. Infect. 20 (Suppl 5), 66–75, doi:10.1111/1469-0691.12570 (2014).
Cheung, G. Y., Wang, R., Khan, B. A., Sturdevant, D. E. & Otto, M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun. 79, 1927–1935, doi:10.1128/IAI.00046-11 (2011).
Montgomery, C. P., Boyle-Vavra, S. & Daum, R. S. Importance of the global regulators agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One 5, e15177, doi:10.1371/journal.pone.0015177 (2010).
Novick, R. P. & Geisinger, E. Quorum sensing in staphylococci. Annu. Rev. Genet. 42, 541–564, doi:10.1146/annurev.genet.42.110807.091640 (2008).
Thoendel, M., Kavanaugh, J. S., Flack, C. E. & Horswill, A. R. Peptide signaling in the staphylococci. Chem. Rev. 111, 117–151, doi:10.1021/cr100370n (2011).
Kaufmann, G. F., Park, J. & Janda, K. D. Bacterial quorum sensing: a new target for anti-infective immunotherapy. Expert Opin. Biol. Ther. 8, 719–724, doi:10.1517/14712598.8.6.719 (2008).
Park, J. et al. Infection control by antibody disruption of bacterial quorum sensing signaling. Chem. Biol. 14, 1119–1127, doi:10.1016/j.chembiol.2007.08.013 (2007).
O’Rourke, J. P. et al. Development of a mimotope vaccine targeting the Staphylococcus aureus quorum sensing pathway. PLoS One 9, e111198, doi:10.1371/journal.pone.0111198 (2014).
Jarraud, S. et al. Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (Alleles), and human disease. Infect. Immun 70, 631–641, doi:10.1128/Iai.70.2.631-641.2002 (2002).
Traber, K. E. et al. agr function in clinical Staphylococcus aureus isolates. Microbiology 154, 2265–2274, doi:10.1099/mic.0.2007/011874-0 (2008).
Kaufmann, G. F., Park, J., Mayorov, A. V., Kubitz, D. M. & Janda, K. D. Generation of quorum quenching antibodies. Methods Mol. Biol 692, 299–311, doi:10.1007/978-1-60761-971-0_22 (2011).
Chackerian, B. Virus-like particles: flexible platforms for vaccine development. Expert review of vaccines 6, 381–390 (2007).
Caldeira, J. C. & Peabody, D. S. Thermal stability of RNA phage virus-like particles displaying foreign peptides. Journal of nanobiotechnology 9, 22 (2011).
Caldeira, J. C. & Peabody, D. S. Stability and assembly in vitro of bacteriophage PP7 virus-like particles. J Nanobiotechnology 5, 10, doi:10.1186/1477-3155-5-10 (2007).
Caldeira Jdo, C. et al. Immunogenic display of diverse peptides, including a broadly cross-type neutralizing human papillomavirus L2 epitope, on virus-like particles of the RNA bacteriophage PP7. Vaccine 28, 4384–4393, doi:10.1016/j.vaccine.2010.04.049 (2010).
Chao, J. A., Patskovsky, Y., Almo, S. C. & Singer, R. H. Structural basis for the coevolution of a viral RNA-protein complex. Nat. Struct. Mol. Biol. 15, 103–105, doi:10.1038/nsmb1327 (2008).
Wright, J. S. 3rd, Jin, R. & Novick, R. P. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc. Natl. Acad. Sci. USA 102, 1691–1696, doi:10.1073/pnas.0407661102 (2005).
Peterson, M. M. et al. Apolipoprotein B Is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe 4, 555–566, doi:10.1016/j.chom.2008.10.001 (2008).
Sully, E. K. et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 10, e1004174, doi:10.1371/journal.ppat.1004174 (2014).
Hall, P. R. et al. Nox2 modification of LDL is essential for optimal apolipoprotein B-mediated control of agr type III Staphylococcus aureus quorum-sensing. PLoS Pathog 9, e1003166, doi:10.1371/journal.ppat.1003166 (2013).
Daly, S. M. et al. Omega-Hydroxyemodin limits Staphylococcus aureus quorum sensing-mediated pathogenesis and inflammation. Antimicrob. Agents Chemother. 59, 2223–2235, doi:10.1128/AAC.04564-14 (2015).
Gray, B., Hall, P. & Gresham, H. Targeting agr- and agr-Like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensors 13, 5130–5166, doi:10.3390/s130405130 (2013).
Tumban, E., Peabody, J., Peabody, D. S. & Chackerian, B. A pan-HPV vaccine based on bacteriophage PP7 VLPs displaying broadly cross-neutralizing epitopes from the HPV minor capsid protein, L2. PLoS One 6 (2011).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 40, W294–297, doi:10.1093/nar/gks493 (2012).
Park, H., Lee, G. R., Heo, L. & Seok, C. Protein loop modeling using a new hybrid energy function and its application to modeling in inaccurate structural environments. PLoS One 9, e113811, doi:10.1371/journal.pone.0113811 (2014).
Tars, K., Fridborg, K., Bundule, M. & Liljas, L. The three-dimensional structure of bacteriophage PP7 from Pseudomonas aeruginosa at 3.7-A resolution. Virology 272, 331–337, doi:10.1006/viro.2000.0373 (2000).
Tars, K., Fridborg, K., Bundule, M. & Liljas, L. Structure determination of bacteriophage PP7 from Pseudomonas aeruginosa: from poor data to a good map. Acta Crystallogr. D Biol. Crystallogr 56, 398–405 (2000).
Tumban, E., Peabody, J., Peabody, D. S. & Chackerian, B. A universal virus-like particle-based vaccine for human papillomavirus: longevity of protection and role of endogenous and exogenous adjuvants. Vaccine 31, 4647–4654 (2013).
Tumban, E., Peabody, J., Tyler, M., Peabody, D. S. & Chackerian, B. VLPs displaying a single L2 epitope induce broadly cross-neutralizing antibodies against human papillomavirus. PLoS One 7, e49751, doi:10.1371/journal.pone.0049751 (2012).
Carrel, M., Perencevich, E. N. & David, M. Z. USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000-2013. Emerg. Infect. Dis. 21, 1973–1980, doi:10.3201/eid2111.150452 (2015).
Rynda-Apple, A. et al. Virus-like particle-induced protection against MRSA pneumonia is dependent on IL-13 and enhancement of phagocyte function. The American journal of pathology 181, 196–210 (2012).
Malachowa, N., Kobayashi, S. D., Braughton, K. R. & DeLeo, F. R. Mouse model of Staphylococcus aureus skin infection. Methods Mol. Biol 1031, 109–116, doi:10.1007/978-1-62703-481-4_14 (2013).
Public health dispatch: outbreaks of community-associated methicillin-resistant Staphylococcus aureus skin infections–Los Angeles County, California, 2002–2003. Can. Commun. Dis. Rep. 29, 110–112 (2003).
Inoshima, N., Wang, Y. & Bubeck Wardenburg, J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Invest. Dermatol. 132, 1513–1516, doi:10.1038/jid.2011.462 (2012).
Kennedy, A. D. et al. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 202, 1050–1058, doi:10.1086/656043 (2010).
Sampedro, G. R. et al. Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J Infect Dis 210, 1012–1018, doi:10.1093/infdis/jiu223 (2014).
Kobayashi, S. D. et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis 204, 937–941, doi:10.1093/infdis/jir441 (2011).
Berube, B. J. & Bubeck Wardenburg, J. Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel) 5, 1140–1166 (2013).
NIAID Antimicrobial Resistance Program: Current Status and Future Directions 2014 - ARstrategicplan2014.pdf, http://www.ncbi.nlm.nih.gov/pubmed/ (2015).
Spellberg, B., Bartlett, J. G. & Gilbert, D. N. The future of antibiotics and resistance. N. Engl. J. Med. 368, 299–302, doi:10.1056/NEJMp1215093 (2013).
DeLeo, F. R., Diep, B. A. & Otto, M. Host defense and pathogenesis in Staphylococcus aureus infections. Infect. Dis. Clin. North Am. 23, 17–34 (2009).
Cheung, G. Y. & Otto, M. The potential use of toxin antibodies as a strategy for controlling acute Staphylococcus aureus infections. Expert Opin. Ther. Targets 16, 601–612 (2012).
Tkaczyk, C. et al. Staphylococcus aureus alpha toxin suppresses effective innate and adaptive immune responses in a murine dermonecrosis model. PLoS One 8, e75103, doi:10.1371/journal.pone.0075103 (2013).
Proctor, R. A. Recent developments for Staphylococcus aureus vaccines: clinical and basic science challenges. European cells & materials 30, 315–326 (2015).
Berube, B. J. & Wardenburg, J. B. Staphylococcus aureus alpha-Toxin: Nearly a Century of Intrigue. Toxins (Basel) 5, 1140–1166, doi:10.3390/toxins5061140 (2013).
Bubeck Wardenburg, J. & Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205, 287–294, doi:10.1084/jem.20072208 (2008).
Adhikari, R. P. et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: protection against bacteremia and pneumonia. PLoS One 7, e38567, doi:10.1371/journal.pone.0038567 (2012).
Oscherwitz, J., Munoz-Planillo, R., Yu, F., Nunez, G. & Cease, K. B. In vivo mapping of a protective linear neutralizing epitope at the N-terminus of alpha hemolysin from Staphylococcus aureus. Mol. Immunol. 60, 62–71 (2014).
Oscherwitz, J. & Cease, K. B. Identification and validation of a linear protective neutralizing epitope in the beta-pore domain of alpha toxin. PLoS One 10, e0116882, doi:10.1371/journal.pone.0116882 (2015).
Yu, X. Q. et al. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, ExtendedHalf-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother doi:10.1128/aac.01020-16 (2016).
Figueroa, M. et al. Polyhydroxyanthraquinones as Quorum Sensing Inhibitors from the Guttates of Penicillium restrictum and Their Analysis by Desorption Electrospray Ionization Mass Spectrometry. J. Nat. Prod 77, 1351–1358, doi:10.1021/Np5000704 (2014).
Khodaverdian, V. et al. Discovery of Antivirulence Agents against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 57, 3645–3652 (2013).
Kuo, D. et al. Novel quorum-quenching agents promote methicillin-resistant Staphylococcus aureus (MRSA) wound healing and sensitize MRSA to beta-lactam antibiotics. Antimicrob. Agents Chemother. 59, 1512–1518, doi:10.1128/AAC.04767-14 (2015).
Yu, G., Kuo, D., Shoham, M. & Viswanathan, R. Combinatorial synthesis and in vitro evaluation of a biaryl hydroxyketone library as antivirulence agents against MRSA. ACS combinatorial science 16, 85–91, doi:10.1021/co400142t (2014).
Cech, N. B., Junio, H. A., Ackermann, L. W., Kavanaugh, J. S. & Horswill, A. R. Quorum quenching and antimicrobial activity of goldenseal (Hydrastis canadensis) against methicillin-resistant Staphylococcus aureus (MRSA). Planta Med. 78, 1556–1561, doi:10.1055/s-0032-1315042 (2012).
Quave, C. L. et al. Castanea sativa (European Chestnut) Leaf Extracts Rich in Ursene and Oleanene Derivatives Block Staphylococcus aureus Virulence and Pathogenesis without Detectable Resistance. PLoS One 10, e0136486, doi:10.1371/journal.pone.0136486 (2015).
Vermote, A. et al. Hamamelitannin Analogues that Modulate Quorum Sensing as Potentiators of Antibiotics against Staphylococcus aureus. Angew. Chem. Int. Ed. Engl. 55, 6551–6555, doi:10.1002/anie.201601973 (2016).
Nakayama, J. et al. Ambuic acid inhibits the biosynthesis of cyclic peptide quormones in gram-positive bacteria. Antimicrob. Agents Chemother. 53, 580–586, doi:10.1128/AAC.00995-08 (2009).
Tal-Gan, Y., Stacy, D. M., Foegen, M. K., Koenig, D. W. & Blackwell, H. E. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J. Am. Chem. Soc. 135, 7869–7882, doi:10.1021/ja3112115 (2013).
Tal-Gan, Y., Ivancic, M., Cornilescu, G., Yang, T. & Blackwell, H. E. Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus. Angew. Chem. Int. Ed. Engl. 55, 8913–8917, doi:10.1002/anie.201602974 (2016).
Gordon, C. P., Williams, P. & Chan, W. C. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J. Med. Chem. 56, 1389–1404 (2013).
Murray, E. J. et al. Targeting Staphylococcus aureus quorum sensing with nonpeptidic small molecule inhibitors. J. Med. Chem. 57, 2813–2819, doi:10.1021/jm500215s (2014).
Chan, W. C., Coyle, B. J. & Williams, P. Virulence regulation and quorum sensing in staphylococcal infections: competitive AgrC antagonists as quorum sensing inhibitors. J. Med. Chem. 47, 4633–4641, doi:10.1021/jm0400754 (2004).
Kirchdoerfer, R. N. et al. Structural basis for ligand recognition and discrimination of a quorum-quenching antibody. J Biol Chem 286, 17351–17358, doi:10.1074/jbc.M111.231258 (2011).
Freitag, N. E., Port, G. C. & Miner, M. D. Listeria monocytogenes - from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 7, 623–628, doi:10.1038/nrmicro2171 (2009).
Garmyn, D., Gal, L., Lemaitre, J. P., Hartmann, A. & Piveteau, P. Communication and autoinduction in the species Listeria monocytogenes: A central role for the agr system. Commun. Integr. Biol 2, 371–374 (2009).
Autret, N., Raynaud, C., Dubail, I., Berche, P. & Charbit, A. Identification of the agr locus of Listeria monocytogenes: role in bacterial virulence. Infect. Immun. 71, 4463–4471 (2003).
Riedel, C. U. et al. AgrD-dependent quorum sensing affects biofilm formation, invasion, virulence and global gene expression profiles in Listeria monocytogenes. Mol. Microbiol 71, 1177–1189, doi:10.1111/j.1365-2958.2008.06589.x (2009).
Rieu, A., Weidmann, S., Garmyn, D., Piveteau, P. & Guzzo, J. Agr system of Listeria monocytogenes EGD-e: role in adherence and differential expression pattern. Appl. Environ. Microbiol. 73, 6125–6133, doi:10.1128/aem.00608-07 (2007).
Rieu, A. et al. Listeria monocytogenes EGD-e biofilms: no mushrooms but a network of knitted chains. Appl. Environ. Microbiol. 74, 4491–4497, doi:10.1128/aem.00255-08 (2008).
Zetzmann, M., Sanchez-Kopper, A., Waidmann, M. S., Blombach, B. & Riedel, C. U. Identification of the agr Peptide of Listeria monocytogenes. Front. Microbiol 7, 989, doi:10.3389/fmicb.2016.00989 (2016).
Kristich, C.J., Rice, L.B. & Arias, C.A. Enterococcal Infection - Treatment and Antibiotic Resistance In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet] (ed. Gilmore, M. S., Clewell, D. B., Ike, Y. & Shankar, N.) (Massachusetts Eye and Ear Infirmary, 2014).
Nakayama, J. et al. Gelatinase biosynthesis-activating pheromone: a peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol. Microbiol 41, 145–154 (2001).
Nakayama, J. et al. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheromone propeptide corresponding to staphylococcal AgrD. J. Bacteriol. 188, 8321–8326, doi:10.1128/jb.00865-06 (2006).
Cook, L. C. & Federle, M. J. Peptide pheromone signaling in Streptococcus and Enterococcus. FEMS Microbiol. Rev. 38, 473–492, doi:10.1111/1574-6976.12046 (2014).
Hancock, L. E. & Perego, M. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 186, 5629–5639, doi:10.1128/jb.186.17.5629-5639.2004 (2004).
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. Effects of Enterococcus faecalis fsr Genes on Production of Gelatinase and a Serine Protease and Virulence, doi:10.1128/IAI.68.5.2579-2586.2000 (2000).
Thurlow, L. R. et al. Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect. Immun. 78, 4936–4943, doi:10.1128/iai.01118-09 (2010).
Engelbert, M., Mylonakis, E., Ausubel, F. M., Calderwood, S. B. & Gilmore, M. S. Contribution of gelatinase, serine protease, and fsr to the pathogenesis of Enterococcus faecalis endophthalmitis. Infect. Immun. 72, 3628–3633, doi:10.1128/iai.72.6.3628-3633.2004 (2004).
Shankar, J., Walker, R. G., Ward, D. & Horsburgh, M. J. The Enterococcus faecalis exoproteome: Identification and temporal regulation by fsr. PLoS One. 7, e33450, doi:10.1371/journal.pone.0033450 (2012).
Darkoh, C., DuPont, H. L., Norris, S. J. & Kaplan, H. B. Toxin synthesis by Clostridium difficile is regulated through quorum signaling. mBio 6, e02569, doi:10.1128/mBio.02569-14 (2015).
Darkoh, C., Odo, C. & DuPont, H. L. Accessory Gene Regulator-1 Locus Is Essential for Virulence and Pathogenesis of Clostridium difficile. mBio 7, doi:10.1128/mBio.01237-16 (2016).
Kuehne, S. A. et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713, doi:10.1038/nature09397 (2010).
Cohen, S. H. et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31, 431–455, doi:10.1086/651706 (2010).
Zhu, J. & Kaufmann, G. F. Quo vadis quorum quenching? Curr. Opin. Pharmacol. 13, 688–698, doi:10.1016/j.coph.2013.07.003 (2013).
Otto, M. Quorum-sensing control in Staphylococci – a target for antimicrobial drug therapy? FEMS Microbiol. Lett 241, 135–141, doi:10.1016/j.femsle.2004.11.016 (2004).
Beenken, K. E. et al. Epistatic relationships between sarA and agr In Staphylococcus aureus biofilm formation. PLoS One 5, e10790, doi:10.1371/journal.pone.0010790 (2010).
Beenken, K. E., Blevins, J. S. & Smeltzer, M. S. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect. Immun. 71, 4206–4211 (2003).
Vuong, C., Saenz, H. L., Gotz, F. & Otto, M. Impact of the agr quorum-sensing system on adherence to polystyrene in Staphylococcus aureus. J Infect Dis 182, 1688–1693, doi:10.1086/317606 (2000).
Saravia-Otten, P., Muller, H. P. & Arvidson, S. Transcription of Staphylococcus aureus fibronectin binding protein genes is negatively regulated by agr and an agr-independent mechanism. J. Bacteriol. 179, 5259–5263 (1997).
Le, K. Y., Dastgheyb, S., Ho, T. V. & Otto, M. Molecular determinants of staphylococcal biofilm dispersal and structuring. Frontiers in cellular and infection microbiology 4, 167, doi:10.3389/fcimb.2014.00167 (2014).
Kavanaugh, J. S. & Horswill, A. R. Impact of Environmental Cues on Staphylococcal Quorum Sensing and Biofilm Development. J Biol Chem 291, 12556–12564, doi:10.1074/jbc.R116.722710 (2016).
Paharik, A. E. & Horswill, A. R. The Staphylococcal Biofilm: Adhesins, Regulation, and Host Response. Microbiology spectrum 4, doi:10.1128/microbiolspec.VMBF-0022-2015 (2016).
Scherr, T. D. et al. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. mBio 6, doi:10.1128/mBio.01021-15 (2015).
Fowler, V. G. Jr. et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 190, 1140–1149, doi:10.1086/423145 (2004).
Sakoulas, G. et al. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Antimicrob. Agents Chemother. 46, 1492–1502 (2002).
Pynnonen, M., Stephenson, R. E., Schwartz, K., Hernandez, M. & Boles, B. R. Hemoglobin promotes Staphylococcus aureus nasal colonization. PLoS Pathog. 7, e1002104, doi:10.1371/journal.ppat.1002104 (2011).
Rothfork, J. M. et al. Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: new role for the NADPH oxidase in host defense. Proc. Natl. Acad. Sci. USA 101, 13867–13872, doi:10.1073/pnas.0402996101 (2004).
Naskalska, A. & Pyrc, K. Virus Like Particles as Immunogens and Universal Nanocarriers. Pol. J. Microbiol. 64, 3–13 (2015).
Lacson, E. et al. Antibody response to Engerix-B and Recombivax-HB hepatitis B vaccination in end-stage renal disease. Hemodialysis international. International Symposium on Home Hemodialysis 9, 367–375, doi:10.1111/j.1492-7535.2005.01155.x (2005).
GARDASIL®9 (Human Papillomavirus 9-valent Vaccine, Recombinant) for Health Care Professionals, https://www.merckvaccines.com/Products/Gardasil9pgid=UoXun1CIyLRSR0EK44UuV0Tn0000rKPQB0Nasid=cz3-ITHtiBPkIWaMzyyb3i5Babw10BqEolE= (2016).
Chackerian, B., Durfee, M. R. & Schiller, J. T. Virus-like display of a neo-self antigen reverses B cell anergy in a B cell receptor transgenic mouse model. J Immunol 180, 5816–5825 (2008).
Chackerian, B., Lowy, D. R. & Schiller, J. T. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J. Clin. Invest 108, 415–423 (2001).
Chackerian, B., Lowy, D. R. & Schiller, J. T. Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. USA 96, 2373–2378 (1999).
Frietze, K. M., Peabody, D. S. & Chackerian, B. Engineering virus-like particles as vaccine platforms. Curr. Opin. Virol 18, 44–49, doi:10.1016/j.coviro.2016.03.001 (2016).
Effio, C. L. & Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnology journal 10, 715–727, doi:10.1002/biot.201400392 (2015).
Bachmann, M. F. & Jennings, G. T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nature reviews.Immunology 10, 787–796 (2010).
Jennings, G. T. & Bachmann, M. F. Immunodrugs: therapeutic VLP-based vaccines for chronic diseases. Annu. Rev. Pharmacol. Toxicol. 49, 303–326 (2009).
Rivera-Hernandez, T. et al. Self-adjuvanting modular virus-like particles for mucosal vaccination against group A streptococcus (GAS). Vaccine 31, 1950–1955, doi:10.1016/j.vaccine.2013.02.013 (2013).
Seth, A. et al. Modular virus-like particles for sublingual vaccination against group A streptococcus. Vaccine, doi:10.1016/j.vaccine.2016.11.008 (2016).
Tamborrini, M. et al. A Synthetic Virus-Like Particle Streptococcal Vaccine Candidate Using B-Cell Epitopes from the Proline-Rich Region of Pneumococcal Surface Protein A. Vaccines 3, 850–874, doi:10.3390/vaccines3040850 (2015).
Alksne, L. E. & Projan, S. J. Bacterial virulence as a target for antimicrobial chemotherapy. Curr. Opin. Biotechnol. 11, 625–636, doi:10.1016/S0958-1669(00)00155-5 (2000).
in National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals (National Academies Press (US), 2011).
Rothfork, J. M., Dessus-Babus, S., Wamel, W. J. V., Cheung, A. L. & Gresham, H. D. Fibrinogen depletion attenuates Staphyloccocus aureus infection by preventing density-dependent virulence gene up-regulation. J Immunol 171, 5389–5395 (2003).
Hjelle, B. L. et al. Methods for Screening Viral Like Particles and Identifying Neutralizing Epitopes and Related Vaccines, Constructs, and Libraries, https://google.com/patents/US20130149336 (2013).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Me thods 9, 671–675 (2012).
Acknowledgements
This work was supported by the National Institutes of Health grants AI091917 and AI114706 (P.R.H.) and AI083305 (BC). S.M.D. was supported by the University of New Mexico Infectious Diseases and Inflammation Training Grant T32-AI007538. Also, we thank Dr. Hattie Gresham for valuable insight.
Author information
Authors and Affiliations
Contributions
S.M.D., D.S.P., B.C. and P.R.H. conceived the experiments; D.S.P. and B.C. provided essential materials; S.M.D., J.A.J., K.D.T., B.O.E., S.P. and K.M.F. carried out the experiments; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Daly, S.M., Joyner, J.A., Triplett, K.D. et al. VLP-based vaccine induces immune control of Staphylococcus aureus virulence regulation. Sci Rep 7, 637 (2017). https://doi.org/10.1038/s41598-017-00753-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00753-0
This article is cited by
-
The Application Potential of Synthetic Biology in Microbial Communication
Current Clinical Microbiology Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
