
Abstract
Node positive head and neck squamous cell carcinomas (HNSCCs) patients exhibit worse outcomes in terms of regional neck control, risk for distant metastases and overall survival. Smaller non-palpable lymph nodes may be inflammatory or may harbor clinically occult metastases, a characterization that can be challenging to make using routine imaging modalities. Ferumoxytol has been previously investigated as an intra-tumoral contrast agent for magnetic resonance imaging (MRI) for intracranial malignancies and lymph node agent in prostate cancer. Hence, our group was motivated to carry out a prospective feasibility study to assess the feasibility of ferumoxytol dynamic contrast enhanced (DCE)-weighted MRI relative to that of gadolinium-based DCE-MRI for nodal and primary tumor imaging in patients with biopsy-proven node-positive HNSCC or melanoma. Although this institutional review board (IRB)-approved study was prematurely terminated because of an FDA black box warning, the investigators sought to curate and publish this unique dataset of matched clinical, and anatomical and DCE MRI data for the enrolled five patients to be available for scientists interested in molecular imaging.
Measurement(s) | imaging assay • head and neck squamous cell carcinoma |
Technology Type(s) | magnetic resonance imaging |
Factor Type(s) | contrast agent |
Sample Characteristic - Organism | Homo sapiens |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.11409516
Similar content being viewed by others


Background & Summary
Neck nodal involvement in head and neck squamous cell carcinomas (HNSCCs) represents one of the most important features for head and neck malignancies risk stratification1. Pretreatment diagnosis of the neck disease now relies on computed tomography (CT) and positron-emission tomography fused with CT (PET/CT) that have many limitations leading to uncertainty and the potential for both over- and under- treatment of patients2. Given the current limitations of conventional imaging, enhanced imaging technologies are relevant to solve this unmet need. Recent data suggest that primary tumor assessment with dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) may reveal quantitative assessments of the tumor environment that are germane to treatment selection and response evaluation3. Currently, gadolinium is the standard of care contrast agent for DCE-MRI4. However, a large array of potential MR contrast agents exists, including ultra-small superparamagnetic iron oxides (USPIO). The USPIO contrast-enhanced MR imaging has been previously shown to be promising as an avenue for differentiation of benign and malignant lymph nodes5,6.
Although a prospective trial in prostate cancer was successfully conducted using a dextran coated USPIO, also known as ferumoxtran-10 (Combidex, AMAG Pharmaceuticals, Inc. Lexington, MA), a Food and Drug Administration (FDA) Advisory Panel recommended against approval of this agent7. Afterwards, a derivative of ferumoxtran-10, ferumoxytol (Feraheme, AMAG Pharmaceuticals, Inc. Lexington, MA, US) was FDA-approved for iron replacement therapy in iron deficiency anemia in chronic kidney disease8,9. Ferumoxytol is a semi-synthetic, carbohydrate-coated, magnetic iron oxide preparation. This compound is taken up by normal lymph nodes and excluded from malignant nodal tissue10.
To date, no USPIO contrast agents were FDA-approved for the indication of head and neck imaging. More interestingly, no prospective data existed regarding ferumoxytol for evaluation of lymph node involvement in HNSCC compared to other imaging methodologies. Hence, our group was motivated to carry out a feasibility study of USPIO DCE-weighted MRI for primary and nodal tumor in locally advanced HNSCCs relative to that of gadolinium-based DCE-MRI. Additionally, it was our aim to investigate the optimum dose, timing, or sequence parameters of MR tumor imaging in HNSCC or melanoma with ferumoxytol.
However, during the course of this IRB-approved study, after 5 patients had enrolled and received MRIs as specified, with no demonstrable toxicities observed, a series of 79 cases of anaphylactic reactions associated with commercial ferumoxytol administration in its listed FDA indication for iron deficiency anemia were identified using the FDA adverse event reporting system database. Patients’ ages ranged from 19 to 96, and almost 50% of all cases reported the anaphylactic reactions occurring with the first dose of ferumoxytol. Of the 79 cases, 18 were fatal despite immediate medical intervention and emergency resuscitation attempts. Consequently, given the potential risk to patients in a Phase 0/1 feasibility trial designed to minimize risk, the investigators discontinued patient accrual and study conduction pursuant to the FDA black box warning against ferumoxytol issued on March 30, 201511.
Notably, however, in the interval since closing the trial, a randomized safety assessment for the FDA approved indication showed less than 1% incidence of moderate‐to‐severe toxicities (including anaphylaxis, or moderate‐to‐severe hypotension) within 5 weeks of ferumoxytol administration12; an update of 217 consecutive patients with wide range of ages, comorbid conditions and kidney functions given ferumoxytol specifically for MRI applications with no observed adverse events13; and a report of 69 patients receiving 85 ferumoxytol injections for MRI with 4 reported mild adverse events (2 episodes mild hypotension and one episode of nausea, and one episode warmness/erythema at the in injection site)14.
Consequently, as more recent enthusiasm for ferumoxytol as a potential (cancer) imaging agent has resurfaced, the potential utility of our extant, if limited, series prompts now public dissemination of the extant data from this terminated trial, and we have opted to curate and publish this unique dataset of matched clinical and imaging data for these 5 patients15. To our knowledge, this is the first and only extant matched USPIO contrast-enhanced MRI scans –among other structural sequences- and clinical data of HNSCC patients where scans were acquired serially prior to radiotherapy (RT) course. This dataset represents a unique repository resource for scientists interested in molecular imaging of underlying tumor biology for other USPIO molecules, as well as those who may find capacity for additional development in HNSCC for ferumoxytol for similar applications.
Methods
Study conception and initiation
Then-current leadership of the licensing vendor for Feraheme has explicitly stated during study development in 2011–2012, that the vendor has no intent or desire to investigate imaging applications in vivo. For this reason, Ed Neuwelt, MD at Oregon Health and Science University acquired orphan drug designation for Ferumoxytol for use in MRI imaging of brain cancers (https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=338411). At the time of protocol approval, ferumoxytol had an excellent safety profile, and based on these rationales, MD Anderson Cancer Center approved regulatory exemption from the Investigational New Drug regulations, 21 CFR 312; as:
(1) The investigational drug is lawfully marketed in the United States; (2) The investigation [was] not intended to be reported to the FDA as a well-controlled study in support of a new indication for use of the drug product, but rather as a feasibility study; (3) The investigation [was] not intended to support a significant change in advertising to an existing lawfully marketed prescription drug product; (4) The investigation does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product; (5) The investigation [was] conducted in compliance with the requirements for institutional review set forth in FDA regulations 21 CFR 56, and requirements for informed consent as set forth in FDA regulations 21 CFR 50; 6) The investigation [was] conducted in compliance with FDA regulations 21 CFR 312.7: Promotion and charging for investigational drugs.
After approval, the protocol was initiated, activated for enrollment, posted on ClinicalTrials.Gov [ClinicalTrials.gov Identifier NCT01895829 (https://clinicaltrials.gov/ct2/show/NCT01895829)]16, and accrual commenced.
Study population and eligibility criteria
This single institute feasibility study was planned to include untreated biopsy-proven HNSCC or melanoma with multiple malignant lymph nodes by imaging (clinical N2b or greater per the American Joint Committee on Caner (AJCC) and Union for International Cancer Control (UICC) cancer staging system, 7th edition17 presenting at MD Anderson Cancer Center This study was approved by the MD Anderson clinical ethics committee. Written informed consents were signed by all participating patients. This trial was also registered in MD Anderson Cancer Center Clinical Trials database as Study #2012–1127.
Inclusion criteria included patients 18 years of age or older with histologically or cytologically confirmed HNSCC or melanoma; along with measurable clinical and/or radiographic poly-nodal disease defined as stage N2b, N2c or N3 disease with multiple involved lymph nodes per the AJCC cancer staging criteria. All patients received or were dispositioned to receive a PET/CT scan within two weeks of starting definitive therapy for their head and neck malignancy and their participation in this study.
Patients were excluded from the study if they have already undergone definitive resection, chemotherapy, or radiation treatment of their primary or nodal disease. Patients were also excluded if they did not give written, informed consent, were unable to undergo MR imaging, incapable of tolerating DCE-MRI, or having an estimated glomerular filtration rate (GFR) < 60 ml/min/1.73 m2. A key exclusion criterion was contraindications to iron supplementation, as hemochromatosis, colitis, history of GI bleeds, alcoholism, or liver disease. Patients that had iron overload and/or hypersensitivity to Feraheme were also excluded. Women of childbearing potential were also ruled out. Male patients were obligated to practice effective contraception throughout the study. Other exclusion criteria were the presence of any evidence of iron overload on pre-imaging laboratory studies, or any contraindications to gadolinium-based contrast agents or claustrophobia.
All patients were clinically evaluated and staged in the standard fashion via physical examination and routine radiographic studies including all of the following: contrast-enhanced CT of the head and neck, PET/CT of the head and neck and chest x-ray. Patients also underwent routine baseline assessment by Dental Oncology, Speech Pathology and Dietary Services. Inclusion and exclusion criteria are summarized as follows:
Inclusion criteria:
-
Patients 18 years of age or older
-
Histologically or cytologically confirmed head and neck squamous cell carcinoma or melanoma
-
Measurable clinical and/or radiographic poly-nodal disease defined as stage N2b, N2c or N3 disease with multiple involved lymph nodes as defined by the AJCC cancer staging criteria
-
Patients who have received or are dispositioned to receive a PET/CT scan within two weeks of starting definitive therapy for their head and neck malignancy and their participation in this study. This implies patients must receive a PET/CT to be eligible for the study.
Exclusion criteria
-
Patients who have undergone definitive resection of their primary or nodal disease as well as any chemotherapy or radiation therapy for their head and neck primary tumor.
-
Patients unable or unwilling to give written, informed consent or to undergo MRI imaging.
-
Women of childbearing potential (A woman of child-bearing potential is a sexually mature woman who has not undergone a hysterectomy or who has not been naturally postmenopausal for at least 24 consecutive months [i.e., who has had menses at any time in the preceding 24 consecutive months]). Male partners must practice effective contraception (oral, injectable, or implantable hormonal contraceptive; tubal ligation; intra-uterine device; barrier contraceptive with spermicide; or vasectomized partner) throughout the study.
-
Patients unable to tolerate DCE-MRI or having an estimated GFR < 60 ml/min/1.73 m2.
-
Contraindications to iron supplementation include hemochromatosis, colitis, and history of GI bleeds, alcoholism, or liver disease. Patients that have iron overload and/or hypersensitivity to Feraheme (brand name for Ferumoxytol), may consequently react negatively to it or anything within it. Consequently, we patients who had symptoms or signs that might be caused by iron overload were excluded. These include patients with (unexplained): arthritis (including premature osteoarthritis), congestive heart failure or cardiomyopathy, adult-onset diabetes, secondary hypogonadism, increased skin pigmentation, or patients with persistently elevated serum ferritin not explained by an underlying inflammatory/systemic disease, unless these patients demonstrate a fasting transferring saturation ≤ 0.45.
-
Patients with any evidence of iron overload on pre-imaging laboratory studies.
-
Patients with any contraindications to gadolinium-based contrast agents.
-
Patients with Claustrophobia.
Patient demographics and clinical end points
Five male patients with oropharyngeal cancer (OPC) and a median age of 67 years (range: 52–78) were included. Patients’ demographic and clinical data are listed in Table 1, whereas Online-only Table 1 contains definitions of the provided clinical data attributes.
The patients’ demographic data encompass age, race or ethnicity, and sex. Disease characteristics include: the OPC subsite, and the TNM (tumor, node, metastasis) status, where the T category describes the size and extent of the initial (primary) tumor, per the AJCC/UICC cancer staging system, 7th edition (25) (https://cancerstaging.org/references-tools/Pages/What-is-Cancer-Staging.aspx). Correspondingly, the N category describes the extent to where the cancer reached the nearby lymph nodes, per the AJCC and UICC cancer staging system, 7th edition, along with the corresponding AJCC stage.
Study design
This study was anticipated as a Phase 0 Feasibility study of USPIO ferumoxytol as a DCE contrast agent in primary and nodal tumor perfusion imaging for HNSCC or melanoma. Twenty patients were originally planned to participate in this study, but only five patients were able to participate and got all scans done before the issuance of the FDA black box warning, and hence the premature closure of the trial. These patients were intended to be accrued to obtain two dynamic susceptibility contrast (DSC) or DCE perfusion MR scans, prior to any chemo or radiotherapy for their primary and nodal disease. At the time of the first scan, patients received the standard non-contrast MRI and a gadolinium-enhanced DCE-weighted MRI per standard institution protocols in immobilization position18. Immobilization entails that patients are unable to move; lying on their back with a bite block, a complete thermoplastic mask, and a posterior tailored head, neck, and shoulder mold for radiotherapy.
Afterwards, following an IV bolus Ferumoxytol injection, T1 and T2-weighted as well as multiparametric MRI scans in immobilization were acquired as Ferumoxytol acts as both a T1 and T2 agent in the tumor19. Later on, patients received a second and a third post-Ferumoxytol scans after various waiting intervals (hours to days) to assess the nodal conspicuity. Exact waiting times relative to initial Ferumoxytol injection are depicted in Table 1.
Safety monitoring included a 45-minute post-Ferumoxytol infusion observation with vital sign monitoring to assess for acute reactions. Serum iron was performed at baseline MRI, 48 hours post-infusion, and 1-month post-infusion. Patients returned for follow-up visits for these toxicity assessments as indicated by the same timeline.
Imaging characteristics and MRI protocol
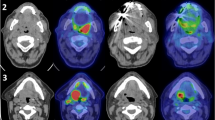
Each patient received 3 anatomical and multi-parametric MRI scans the week before initiating RT. Figure 1 depicts the scanning timeline along with an illustration of how USPIO pattern of enhancement can discriminate benign and malignant lymph nodes. The interval between each scan and the following one ranged between 6 and 96 hours (Table 1). The scans were obtained using a 3.0 T Discovery 750 MRI scanner (GE Healthcare) with a flat insert table (GE Healthcare) and six-element flex coils. The exact same immobilization devices (individualized shoulder and neck mold, tailored head support, and bite block) were utilized in longitudinal scans to enhance co-registration of images and diminish physical motion (e.g., swallowing). This design structure was explained by Hollows et al. where it was shown that this setup with the bite block is much more reproducible than the usual setup20. Although this setup has not been proven to be reproducible yet, this enhanced reproducibility demonstrated at one institution would probably extend to other institutions21.

The scanning timeline and an illustration of pattern of enhancement of iron oxide nanoparticle magnetic resonance imaging.
Thirty slices, 4 mm thick each, with a field of view of 25.6 cm, were chosen to screen the spatial region including the palatine process region cranially to the cricoid cartilage caudally for all scans. Before the DCE-weighted MRI scan, T1 mapping was done by utilizing six variable-flip-angle 3D spoiled gradient recalled echo sequences (SPRG) (flip angles: 2°, 5°, 10°, 15°, 20°, and 25°; repetition time/echo time, 6.1/2.1 ms; effective number of excitations, 0.7 (GE terminology, number of averages = percent sampling * number of signal averages/number of excitations); acquisition resolution, 2 mm × 2 mm × 4 mm; zero filling interpolation, × 2; scan time, 3 min). The DCE-MRI acquisition included a three-dimensional quick spoiled gradient recalled echo sequence to gain enough temporal resolution, contrast, and signal-to-noise ratio (SNR). These scan parameters were employed: a 15° flip angle; a repetition/echo time of 3.6/1.0 ms; 0.7 excitations; an acquisition resolution of 2 mm × 2 mm × 4 mm; a × 2 zero filling interpolation; 5:08 min scanning time; 5.5 s temporal resolution; 50 kHz bandwidth; an acceleration factor of 2; and 56 temporal. The DSC-MRI acquisition included a spin echo EPI sequence to gain enough temporal resolution (1.5 sec), contrast, and signal-to-noise ratio (SNR). These scan parameters were employed: a 45° flip angle; a repetition/echo time of 1500/14.2 ms; an acquisition resolution of 2 mm × 2 mm × 4 mm; 2:15 min scanning time; 1.5 s temporal resolution; 250 kHz bandwidth; an acceleration factor of 2; and 90 temporal.
The DCE/DSC MRI scan was initiated before, during, and following a bolus injection of 0.1 mmol/kg gadopentetate dimeglumine (Magnevist, Bayer Healthcare Pharmaceuticals) delivered at 3 ml/s followed by a 20 ml saline flush also delivered at 3 ml/s. An hour later, subjects received a dose of Ferumoxytol containing 512 mg elemental iron (the FDA package dose for iron deficiency anemia), injected as an IV bolus push as per package insert instructions. A Medrad Solaris MR-compatible power injector was used for all contrast agent injections in order to standardize the contrast agent administration. Scanner and acquisition parameters for various MRI sequences obtained are listed in Online-only Table 2.
Data anonymization
We used the Clinical Trial Processor (CTP), developed by the Radiological Society of North America (RSNA) to anonymize the imaging DICOM files22. Data anonymization was completed in conformation with the HIPAA, as attributed by the DICOM standards committee Attribute Confidentiality Profile (DICOM PS 3.15: Appendix E), which explains the standardized methods and documentation for eradication of secured health records from DICOM images23. A final DICOM anonymization quality assurance was employed utilizing a software, ImageJ (https://imagej.nih.gov/ij/), which compiles all DICOM header tags for an image in a report that we later manually reviewed to ensure that ideal anonymization was achieved. The DICOM files were uploaded to VelocityAI™ 3.0.1 software, where the images were visually examined, to verify that our process of anonymization has not changed the spatial information encompassed in the DICOM header.
Data Records
This head and neck cancer imaging dataset of 5 patients includes anonymized anatomical and multiparametric MRI sequences that are listed in Online-only Table 2. These scans were acquired at three different time points per patient; all prior to the initiation of radiotherapy course. Pertinent clinical meta-data files were also presented as.csv sheets, i.e., files with values separated by commas. This allows data to be put in a structured table format. CSVs are like spreadsheets with a .csv extension. The data records and supplemental descriptions of the clinical meta-data files are cited under figshare (https://doi.org/10.6084/m9.figshare.c.4420499.v2)24. All images followed the standardized DICOM format and the images are labeled by anonymized patient ID numbers. Also, using the identifier, the Patient IDs can be cross-referenced to the data table ‘PatientID’ column.
The imaging dataset is organized into 5 main folders. Each folder name represents individual patient’s ID that starts with ‘USPIO_’ followed by a number; 001 through 005. Under each folder, study participants can locate 3 folders, for all respective time points: baseline, first post-USPIO, and second post-USPIO scans. The next level of subfolders represent various MR sequences acquired at the corresponding time point (Fig. 2). The DICOM tag (0018,1314) classifies the flip angle that was used for the image slice in the variable flip angle images.

The directory structure of uploaded imaging dataset at figshare.
Technical Validation
ClinicStation (Electronic Medical Record System), a custom-built electronic medical record system by MDACC, that started in 1999 with subsequent significant improvement in 2007 that allowed further new capabilities, such as integrating research data and accessing data from virtually every electronic source within the institution, thus serving a great role in patient care and research. http://www.clinfowiki.org/wiki/index.php/ClinicStation.
Usage Notes
We invite all interested researchers to download this dataset of matched clinical-imaging dataset including DCE/DSC- weighted perfusion MRI acquisitions in immobilization position using an under-investigated contrast agent in the setting of HNSCC imaging. Moreover, this data descriptor serves as a report for the only trial that prospectively investigated the feasibility of USPIO as a contrast agent for DCE-MRI acquisition purposes in the setting of head and neck cancer imaging.
Code availability
The Clinical Trial Processor (CTP) that was created by the Radiological Society of North America (RSNA) is available at: https://www.rsna.org/ctp.aspx. The RSNA CTP aligns meticulously to regulations for image anonymization, per the HIPAA Privacy Rule and the DICOM Working Group 18 Supplement 142. Being compatible with all commercially available picture archiving and communication systems (PACS), the RSNA CTP is designed to transport images to online data repositories where the anonymization process takes place. This program replaces patient tags in the DICOM files in a folder (and sub-folders) with anonymized tags that were assigned.
ImageJ, a free software offered by the National Institutes of Health, USA, as a public domain Java processing program. The code for the software is accessible at: https://imagej.nih.gov/ij/.
References
Chen, C. C., Lin, J. C. & Chen, K. W. Lymph node ratio as a prognostic factor in head and neck cancer patients. Radiat Oncol. (London, England) 10, 181 (2015).
Paidpally, V. et al. FDG-PET/CT imaging biomarkers in head and neck squamous cell carcinoma. Imaging Med. 4, 633–647 (2012).
Egeland, T. A. et al. Dynamic contrast-enhanced magnetic resonance imaging of tumors: preclinical validation of parametric images. Radiat Res. 172, 339–347 (2009).
Yan, Y., Sun, X. & Shen, B. Contrast agents in dynamic contrast-enhanced magnetic resonance imaging. Oncotarget. 8(43491), 43505 (2017).
Harada, T., Tanigawa, N., Matsuki, M., Nohara, T. & Narabayashi, I. Evaluation of lymph node metastases of breast cancer using ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging. Eur. JRadio. 63, 401–407 (2007).
Will, O. et al. Diagnostic precision of nanoparticle-enhanced MRI for lymph-node metastases: a meta-analysis. Lancet Oncol. 7, 52–60 (2006).
Heesakkers, R. A. et al. Prostate cancer: detection of lymph node metastases outside the routine surgical area with ferumoxtran-10-enhanced MR imaging. Radiology. 251, 408–414 (2009).
Turkbey, B. et al. A Phase I Dosing Study of Ferumoxytol for MR Lymphography at 3 T in Patients With Prostate Cancer. AJR Am J Roentgenol. 205, 64–69 (2015).
Vasanawala, S. S. et al. Safety and technique of ferumoxytol administration for MRI. Magn Reson Med. 75, 2107–2111 (2016).
Toth, G. B. et al. Current and potential imaging applications of ferumoxytol for magnetic resonance imaging. Kidney Int. 92, 47–66 (2017).
FDA Drug Safety Communication: FDA strengthens warnings and changes prescribing instructions to decrease the risk of serious allergic reactions with anemia drug Feraheme (ferumoxytol), https://www.fda.gov/Drugs/DrugSafety/ucm440138.htm, (U.S. Food and Drug Administration, 2015).
Adkinson, N. F. et al. Comparative safety of intravenous ferumoxytol versus ferric carboxymaltose in iron deficiency anemia: A randomized trial. Am J Hematol. 93, 683–690 (2018).
Nguyen, K.-L. et al. MRI with ferumoxytol: A single center experience of safety across the age spectrum. J Magn Reson Imaging. 45, 804–812 (2017).
Muehe, A. M. et al. Safety Report of Ferumoxytol for Magnetic Resonance Imaging in Children and Young Adults. Invest Radiol. 51, 221–227 (2016).
Toth, G. B. et al. Current and potential imaging applications of ferumoxytol for magnetic resonance imaging. Kidney Int. 92, 47–66 (2017).
M.D. Anderson Cancer Center. Ferumoxytol - Iron Oxide Nanoparticle Magnetic Resonance Dynamic Contrast Enhanced MRI. Identifier NCT01895829 (U.S. National Library of Medicine, 2013).
Edge, S. B. & Compton, C. C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 17, 1471–1474 (2010).
Elhalawani, H. et al. Dynamic contrast-enhanced magnetic resonance imaging for head and neck cancers. Sci Data. 5, 180008 (2018).
Dósa, E. et al. MRI using ferumoxytol improves the visualization of central nervous system vascular malformations. Stroke. 42, 1581–1588 (2011).
Hollows, P., Hayter, J. P. & Vasanthan, S. The Leicester radiotherapy bite block: an aid to head and neck radiotherapy. Br J Oral Maxillofac Surg. 39, 49–51 (2001).
Ger, R. B. et al. A Multi-Institutional Comparison of Dynamic Contrast-Enhanced Magnetic Resonance Imaging Parameter Calculations. Sci Reports. 7, 11185 (2017).
Radiological Society of North America I. CTP-The RSNA Clinical Trial Processor. Radiological Society of North America, Inc.
Fetzer, D. T. & West, O. C. The HIPAA privacy rule and protected health information: implications in research involving DICOM image databases. Acad Radiol. 15, 390–395 (2008).
Elhalawani, H., Mohamed, A. & Fuller, C. Iron Oxide Nanoparticle Dynamic Contrast Enhancement Magnetic Resonance Imaging for Primary and Nodal Tumor in Locally Advanced Head and Neck Tumors: Data from a Terminated Phase 0 Feasibility Study Collection. figshare. https://doi.org/10.6084/m9.figshare.c.4420499.v2 (2019).
Acknowledgements
This study was conducted on behalf of the Joint Head and Neck Radiotherapy-MRI Development Cooperative. Multiple funders/agencies contributed to personnel salaries or project support during the manuscript preparation interval. Dr. Elhalawani is supported in part by the philanthropic donations from the Family of Paul W. Beach to Dr. G. Brandon Gunn, MD. Drs. Elhalawani and Fuller received salary support from National Institutes of Health (NIH)/National Cancer Institute (NCI) Head and Neck Specialized Programs of Research Excellence (SPORE) Developmental Research Program Award (P50 CA097007-10). Drs. Fuller and Mohamed receive(d) salary support from NIH, including: National Institute for Dental and Craniofacial Research Award (1R01DE025248-01/R56DE025248-01); NCI Early Phase Clinical Trials in Imaging and Image-Guided Interventions Program (1R01CA218148-01); NCI Big Data to Knowledge (BD2K) Early Stage Development of Technologies in Biomedical Computing, Informatics, and Big Data Science Award (1R01CA214825-01); NCI Cancer Center Support Grant (CCSG) Radiation Oncology and Cancer Imaging Program Pilot Research Program Award (P30CA016672); NCI/National Science Foundation Joint NSF/NIH Initiative on Quantitative Approaches to Biomedical Big Data (QuBBD) award (5R01CA225190-0); and the National Institute of Biomedical Imaging and Bioengineering (NIBIB) Research Education Program (R25EB025787). This research is supported by the Andrew Sabin Family Foundation; Dr. Fuller is a Sabin Family Foundation Fellow. Dr. Fuller has received direct institutional academic/industry grant support from Elekta AB via an Elekta AB/MD Anderson Department of Radiation Oncology Seed Grant. Dr. Fuller has received speaker travel and honoraria from Elekta AB. The authors would also like to acknowledge the contributions of Drs. Jason Stafford and Edward Jackson towards programmatic oversight and concept approval.
Author information
Authors and Affiliations
Contributions
All listed co-authors performed the following: 1. Substantial contributions to the conception or design of the work. 2. Drafting the work or revising it critically for important intellectual content. 3. Final approval of the version to be published. 4. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Specific additional individual cooperative effort contributions to study/manuscript design/execution/interpretation, in addition to all criteria above are listed as follows: • H.E.: Drafted manuscript; supervised data set anonymization and uploading to data repository. • M.J.A.: Drafted manuscript; assisted with study conception. • Y.D.: Drafted portions of clinical M.R.I. acquisition protocol under supervision; oversight of imaging acquisition. • A.S.R.M.: Study coordinator; assisted with study conception; clinical data collection; prospective patient enrolment and administrative oversight. • A.K.E. and I.A.G.: Assisted with manuscript drafting. • J.W., J.H., G.B.G., S.Y.L., S.J.F., L.E.G. and D.I.R.: Programmatic development, trial coordination assistance and project support; manuscript and study design assistance. • D.I.R.: Responsible for mentored oversight of clinical/research personnel and direct clinical trial development mentorship (C.D.F.). • C.D.F.: Study initiator and director; oversight of all portions of study, including clinical data acquisition and anonymization, patient data collection, pre and post-processing, study coordination, manuscript drafting, manuscript review and approval. Supervision of trainees (A.K.E., I.A.G.) and clinical/research staff (H.E., A.S.R.M., Y.D.).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Tables
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Elhalawani, H., Awan, M.J., Ding, Y. et al. Data from a terminated study on iron oxide nanoparticle magnetic resonance imaging for head and neck tumors. Sci Data 7, 63 (2020). https://doi.org/10.1038/s41597-020-0392-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0392-z
