
Abstract
Dreissenids are established model species for ecological and ecotoxicological studies, since they are sessile and filter feeder organisms and reflect in situ freshwater quality. Despite this strong interest for hydrosystem biomonitoring, omics data are still scarce. In the present study, we achieved full de novo assembly transcriptomes of digestive glands to gain insight into Dreissena polymorpha and D. rostriformis bugensis molecular knowledge. Transcriptomes were obtained by Illumina RNA sequencing of seventy-nine organisms issued from fifteen populations inhabiting sites that exhibits multiple freshwater contamination levels and different hydrosystem topographies (open or closed systems). Based on a recent de novo assembly algorithm, we carried out a complete, quality-checked and annotated transcriptomes. The power of the present study lies in the completeness of transcriptomes gathering multipopulational organisms sequencing and its full availability through an open access interface that gives a friendly and ready-to-use access to data. The use of such data for proteogenomic and targeted biological pathway investigations purpose is promising as they are first full transcriptomes for this two Dreissena species.
Measurement(s) | transcriptome |
Technology Type(s) | RNA sequencing |
Factor Type(s) | sampling location |
Sample Characteristic - Organism | Dreissena polymorpha • Dreissena rostriformis bugensis |
Sample Characteristic - Environment | freshwater biome |
Sample Characteristic - Location | Metropolitan France |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9934226
Similar content being viewed by others
Background & Summary
Dreissena polymorpha and D. rostriformis bugensis, also known as the zebra and the quagga mussels, are model species in ecology and ecotoxicology since the early eighties. These invasive species are now commonly found in freshwaters of the northern hemisphere. D. polymorpha belongs to the 100 of the World’s Worst Invasive Alien Species identified by the IUCN (http://www.iucngisd.org/gisd/100_worst.php). It spread from Ponto-Caspian basin to northern and western Europe in the 19th century, and to North America and Middle East (Turkey) in the late 20th century, while D. r. bugensis colonized both North America and Western Europe between the late 20th and at the beginning of the 21th century1,2. They can reach high densities in invaded ecosystems3, inducing important ecological and economic damages4,5. Their distribution and invasion dynamic is well documented, as well as ecological features of colonized ecosystems. Dreissenids thus become model species in ecology of biological invasion to identify pathway of invasion, and genetic mechanisms associated to colonization at various spatial and temporal scales.
Their ability to tolerate a wide range of environmental contaminants and their presence in almost every major hydrosystem in Europe and North America also lead dreissenids to become a model species for ecotoxicologists, who identified them as the counterpart of Mytilus for freshwaters6. Indeed, being abundant, sessile, filter feeder, mussels bioaccumulate contaminants present in the water column, and are good candidates to be included in biomonitoring programs. It allows to evaluate both the presence of contaminants through bioaccumulation measurements7,8 and their effects through the use of biomarkers, either in the field or in laboratory9,10,11.
A bibliographic research with the keyword “dreissena” performed on the Web of Science the 22th of january 2019 reveals that 3,667 articles were published since 1990, with ca. 180 article each year since 2010. In the meantime, these articles were cited 97,000 times, with a h-index of 119. Their distribution among scientific topics (Fig. 1) reveals this dual interest in ecology and ecotoxicology. However, over the eleven Bioprojects present in NCBI for Dreissena, most focused on foot, gill or mantle tissues where few concern digestive glands. Moreover, the majority of their studies are not de novo RNA-seq but micro-array studies and almost none of them made sequences available in accessible repositories. In the present study, digestive gland was chosen for its importance in ecotoxicology due to its central roles in detoxication functions and energy metabolism12.

Top 10 fields treemap for Dreissena publications.
Indeed, genetic studies on zebra and quagga mussel mainly concerns their spread dynamic through a population genetic perspective13,14,15, the identification of genetic markers for their identification, and bivalve phylogeny and phylogeography13,16,17,18,19. When the focus of the study was to elucidate toxic effect pathways, only a very limited set of genes were targeted, corresponding to the main processes implied in organism’s response to contaminant20,21.
Studies from several populations of the two species can pave the way for several new research possibilities. The most promising and useful ones are to make available a reference transcriptome for proteogenomic studies, to identify sequences to develop new sets of biomarkers, and to better understand acclimation mechanisms occuring during the invasion process and implied in tolerance to contamination.
Methods
Sampling and RNA extraction
Samples were performed on 12 sites representative of french dreissenid populations diversity. Sites were chosen according to their variable contamination levels, chemicophysical quality and topography characteristics (see Supplementary Table 1). Five bivalves were sampled by populations and digestive glands were in situ dissected and stored in RNAlater (Qiagen) at 4 °C. Total RNA extraction was done using RNeasy MinElute kit (Qiagen) according to the manufacturer’s protocol with slight modifications. Briefly, tissus disruption was done first with a pestle and then with a beads beater with glass bead (200 µm) for 1 min. at max speed in 1 mL of RLT buffer supplemented with 40 mM DTT. Samples were centrifuged 3 min at 20,000 × g. Qiagen protocol was used for the next steps. Genomic DNA was removed by DNase digestion on column and total RNA was eluted in ultra pure water. RNA purity and quantity were assessed by OD measurements (OD 260 nm and OD ratio 260/280 and 260/230) and RNA integrity was checked using Bioanalyseur 2100 (Agilent, CA, USA). Total RNA samples were stored at −80. Samples were send to GeT-PlaGe core facility in dried-ice.
RNA sequencing
RNAseq was performed at the GeT-PlaGe core facility, INRA Toulouse. RNA-seq libraries have been prepared according to Illumina’s protocols using the Illumina TruSeq Stranded mRNA sample prep kit to analyze mRNA. The 79 individuals were sequenced separately on 8 different lanes of one flow cell. Briefly, mRNA were selected using poly-T beads. Then, RNA were fragmented to generate double stranded cDNA and adaptators were ligated to be sequenced. Eleven cycles of PCR were applied to amplify libraries. Library quality was assessed using a Fragment Analyser and libraries were quantified by QPCR using the Kapa Library Quantification Kit. RNA-seq experiments have been performed on an Illumina HiSeq3000 using a paired-end read length of 2 × 150 pb with the Illumina HiSeq3000 sequencing kits.
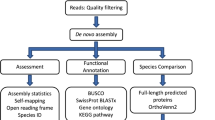
De novo assembly and annotation
The RNA-seq libraries read quality was evaluated using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads from 45 D. polymorpha and 34 D. rostriformis bugensis individuals were cleaned, filtered and de novo assembled for each species with DRAP (De novo RNA-seq Assembly Pipeline, version 1.7)22 using the Oases assembler23. Contigs were kept if they had at least one FPKM. Contigs were then aligned with NCBI BLAST (version 2.2.26, e-value under 1e-5 parameter) on Refseq, Swissprot and three databases issued from Ensembl (Crassostrea gigas, Lottia gigantea and Lingula anatina sequences) to retrieve corresponding annotations. Contigs were also processed with RNAmmer (version 1.2, standard parameters)24 to find ribosomal genes, with RepeatMasker (version open-4-0-3, -engine crossmatch -gccalc -species Crassostrea gigas parameters, http://www.repeatmasker.org) to list contained repeats and with InterProScan (version 4.8, -goterms -pathways parameters)25 for gene ontology and structural annotation. Reads were realigned back to contigs with BWA (version 0.7.12, standard parameters, mem algorithm)26. The resulting sam files were compressed, sorted and indexed with SAMtools (view, sort and index programs, version 1.1, standard parameters)27. Contig expression counts were generated from the bam files with SAMtools (IdxStats program, version 1.1, standard parameters) and merged with UNIX commands (cut, paste). Alignment files were then filtered for duplicates with SAMtools (rmdup program, version 1.1, standard parameters) before variant calling (SNPs and INDELs). Resulting bam files were processed with GATK (version v3.0–0-g6bad1c6, -glm BOTH parameter) following the best practices found on the GATK website28. All the results were uploaded in a RNAbrowse instance29 and can be accessed from the web at http://ngspipelines.toulouse.inra.fr:9014/. Further KEGG annotation were done with KAAS interface (KEGG Automatic Annotation Server) using contig sequences in fasta format.
Biomarker explorations were done using the implemented BLAST in NGSpipeline interface. Biomarker sequences come from close related species (i.e. molluscs). Candidates were selected by the user on score, e-values, identity and length. If several candidates exist, the sequence with highest score was selected.
Data Records
Raw reads and assemblies were gathered in the same NCBI BioProject (PRJNA507340) which includes all BioSamples used for transcriptome assembly (Table 1)30,31,32. All datasets were also available online on sequencing platform web interface (Table 2)33. Datasets annotations (contigs, KEGG, GO) are available on Figshare (Table 3)34.
Technical Validation
Extraction and RNA integrity
Total RNA purity was assessed with a Nanodrop ND-100 Spectrophotometer (Nanodrop Technologies, Wilmington, USA) and RNA with a 260/280 and 260/230 ratio superior to 1.8 were kept. RNA integrity was evaluated with a Bioanalyzer (Agilent RNA 6000 Nano kit). Due to a non conventional 18S/28S ribosomal ratio in bivalve, sample quality was evaluated with the 18S/28S ratio and on the electropherogram (Supplementary Fig. S1). The absence of degradation fragment in the 5S, Fast and Inter regions was a criteria of selection for sequencing.
De novo transcriptome assembly validation
A total of 94,217 contigs were assembled spanning from 200 to 40,000 bp and with an average length of 2,314 bp for D. polymorpha and 1,972 for D. r. bugensis (Table 4). Assembly validation aims at verifying the correspondence between contigs and assembled reads, between contigs and the proteome of a phylogenetically related species and to check if the contigs host single copy awaited proteins. First, reads were mapped back to contigs for each sample in order to monitor the realignment rates revealing a realignment rate above 95% for both species (Table 5). Second, Crassostrea gigas proteins were aligned to contigs using BLAT (v. 35 × 1)35. Proteins mapping at 50% identity and over 50% of the protein length were counted to measure the share of proteins correctly reconstructed by the assembly (Table 6). Last, contigs were processed with BUSCO version 3.0.236 using the metazoa OrthoDB (v. 9) database to check for awaited proteins (Table 7) which shows that almost 95% of expected metazoa BUSCO sequences were found as complete in our de novo transcriptome.
Annotation quality
More than the half of assembled contigs found annotation with an e-value under 1e-5 (59% of 44,538 D. polymorpha contigs and 57% of 49,679 D. rostriformis bugensis contigs). Among top 5 species found as best annotation, Crassostrea gigas represent the best hit species matching with approximately 53% of D. polymorpha and D. r. bugensis sequences (Fig. 2). Among the “others” category, some non bivalves sequences can be present thanks to the analyzed tissue (as digestive gland may contain processing foods). Sequence functional annotations successfully attribute GOterms to 35,4% and KEGG to 21.6% of D. polymorpha contigs and attribute GOterms to 34,2% and KEGG to 20.6% of D. r. bugensis contigs. Full contigs annotation as well as functional annotations are available in the figshare deposit in csv format for quick and easy reuse (Table 3).

Most representated species hits. Top 5 best species hits for (A) Dreissena polymorpha and (B). Dreissena rostriformis bugensis.
Usage Notes
We present here the first whole de novo transcriptomes of the two invasive Dreissena polymorpha and D. rostriformis bugensis species who will be fully available in TSA repository. One of the strength of proposed transcriptome is the conditions in which it was obtained: by investigating individuals coming from several populations, inhabiting contrasted hydrosystems, we encompassed as much as possible the variability of the transcriptome induced by environmental conditions (pollution, biotic interactions, physico-chemistry, climate…), with a mRNA set that is representative of this ecological heterogenity. Our ambitious approach also lead to identification of numerous sequence variants (insertion, deletion and single-nucleotide polymorphism), since studied populations are established for a long time and may have been prone to mutation.
In ecotoxicology, biomarkers implied in responses to oxidative stress, heat shock or xenobiotic exposure are used as indicators of stress. Nowadays, multibiomarker approaches have become a standard, considering that the diversity of contaminants that organisms are exposed to can generate a wide diversity of biological responses. Here, we first wanted to focus on the most frequently used biomarkers in ecotoxicology and which are listed in Table 8. Biomarkers were screened, manually checked and all were found expressed in our both species digestive glands. This table also shows the closest species hits (blastn) and biomarkers selected here exhibit relatively high value of homology with the corresponding sequence in other mollusk, supporting our contig assignations. The high sequence conservation levels observed here may facilitate the development and adaptation of further measurements assays from existing assays in close related species. Interspecies sequence alignments were also investigated indicating a high levels of homologies between the two species (Table 9). In the same way, this may lead to the development of biomarker based assays that, because of sequence similarity, could be cross compatible for D. polymorpha and D. r. bugensis.
By providing Dreissena sequences through a user-friendly interface, we open the way to further explorations of Dreissenids molecular mechanisms by such biomarker assays development, primer design allowing targeted expression analysis or promising proteogenomics studies when coupling with mass spectrometry analysis.
Code availability
Parameters to involved softwares tools are described in the following paragraph.
DRAP (De novo RNA-seq Assembly Pipeline): version 1.7, code available online at http://www.sigenae.org/drap/quick_start.html.
BLAST: version 2.2.26, e-value under 1e-5 parameter.
RNAmmer: version 1.2, standard parameters.
RepeatMasker: version open-4-0-3, -engine crossmatch -gccalc -species
Crassostrea gigas parameters.
InterProScan: version 4.8, -goterms -pathways parameters.
BWA: version 0.7.12, standard parameters, mem algorithm.
SAMtools view, sort and index programs: version 1.1, standard parameters.
SAMtools IdxStats program: version 1.1, standard parameters.
SAMtools rmdup program: version 1.1, standard parameters.
GATK: version v3.0-0-g6bad1c6, -glm BOTH parameter.
BLAT: version 35 × 1, standard parameters.
BUSCO: version 3.0.2 using the metazoa OrthoDB (v. 9), standard parameters.
References
Karatayev, A. Y., Claudi, R. & Lucy, F. E. History of Dreissena research and the ICAIS gateway to aquatic invasions science. Aquat. Invasions 7, 1–5 (2012).
Marescaux, J. et al. Sympatric Dreissena species in the Meuse River: towards a dominance shift from zebra to quagga mussels. Aquat. Invasions. 10, 287–298 (2015).
Akopian, M., Garnier, J., Testard, P. & Ficht, A. Estimating the benthic population of Dreissena polymorpha and its impact in the lower Seine River, France. Estuaries 24, 1003–1014 (2001).
Devin, S., Bollache, L., Noël, P.-Y. & Beisel, J.-N. Patterns of biological invasions in french freshwater systems by non-indigenous macroinvertebrates. Hydrobiologia. 551, 137–146 (2005).
Pimentel, D., Zuniga, R. & Morrison, D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol. Econ. 52, 273–288 (2005).
Binelli, A., Della Torre, C., Magni, S. & Parolini, M. Does zebra mussel (Dreissena polymorpha) represent the freshwater counterpart of Mytilus in ecotoxicological studies? A critical review. Environ. Pollut. 196, 386–403 (2015).
Mills, E. L., Roseman, E. F., Rutzke, M., Gutenmann, W. H. & Lisk, D. J. Contaminant and nutrient element levels in soft tissues of zebra and quagga mussels from waters of southern Lake Ontario. Chemosphere. 27, 1465–1473 (1993).
Bourgeault, A., Gourlay-Francé, C., Priadi, C., Ayrault, S. & Tusseau-Vuillemin, M.-H. Bioavailability of particulate metal to zebra mussels: biodynamic modelling shows that assimilation efficiencies are site specific. Environ. Pollut. 159, 3381–3389 (2011).
De Lafontaine, Y. et al. Biomarkers in zebra mussels (Dreissena polymorpha) for the assessment and monitoring of water quality of the St Lawrence River (Canada). Aquat. Toxicol. 50, 51–71 (2000).
Faria, M. et al. Separating natural from anthropogenic causes of impairment in zebra mussel (Dreissena polymorpha) populations living across a pollution gradient. Aquat. Toxicol. 152, 8295 (2014).
Palais, F. et al. One-year monitoring of core biomarker and digestive enzyme responses in transplanted zebra mussels (Dreissena polymorpha). Ecotoxicology. 21, 888–905 (2012).
Viarengo, A., Lowe, D., Bolognesi, C., Fabbri, E. & Koehler, A. The use of biomarkers in biomonitoring: a 2-tier approach assessing the level of pollutant-induced stress syndrome in sentinel organisms. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 146, 281–300 (2007).
Albrecht, C., Schultheiÿ, R., Kevrekidis, T., Streit, B. & Wilke, T. Invaders or endemics? Molecular phylogenetics, biogeography and systematics of Dreissena in the Balkans. Freshw. Biol 52, 1525–1536 (2007).
Stepien, C. A., Taylor, C. D. & Dabrowska, K. A. Genetic variability and phylogeographical patterns of a nonindigenous species invasion: a comparison of exotic vs native zebra and quagga mussel populations. J. Evol. Biol 15, 314–328 (2002).
Penarrubia, L., Sanz, N., Pla, C., Vidal, O. & Vinas, J. Using massive parallel sequencing for the development, validation, and application of population genetics markers in the invasive bivalve zebra mussel (Dreissena polymorpha). PLoS One. 10, e0120732 (2015).
Gonzalez, V. L. et al. A phylogenetic backbone for bivalvia: an RNA-seq approach. Proc. Biol. Sci. 282, 20142332 (2015).
Soroka, M. et al. Next-generation sequencing of Dreissena polymorpha transcriptome sheds light on its mitochondrial DNA. Hydrobiologia. 810, 255–263 (2018).
Therriault, T. W., Docker, M. F., Orlova, M. I., Heath, D. D. & MacIsaac, H. J. Molecular resolution of the family Dreissenidae (Mollusca: Bivalvia) with emphasis on Ponto-Caspian species, including first report of Mytilopsis leucophaeata in the Black Sea basin. Mol. Phylogenet. Evol. 30, 479–489 (2004).
Geda, S. R. et al. Multilocus phylogeny of the zebra mussel family Dreissenidae (Mollusca: Bivalvia) reveals a fourth neotropical genus sister to all other genera. Mol. Phylogenet. Evol. 127, 1020–1033 (2018).
Hanana, H., Turcotte, P., Andre, C., Gagnon, C. & Gagne, F. Comparative study of the effects of gadolinium chloride and gadolinium - based magnetic resonance imaging contrast agent on freshwater mussel, Dreissena polymorpha. Chemosphere. 181, 197–207 (2017).
Bultelle, F., Panchout, M., Leboulenger, F. & Danger, J. M. Identification of differentially expressed genes in Dreissena polymorpha exposed to contaminants. Mar. Environ. Res. 54, 385–389 (2002).
Cabau, C. et al. Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ. 5, e2988 (2017).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 28, 1086–1092 (2012).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30, 1236–1240 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next- generation DNA sequencing data. Genome res. 20, 1297–1303 (2010).
Mariette, J. et al. RNAbrowse: RNA-Seq de novo assembly results browser. PLoS One. 9, e96821 (2014).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP173947 (2018).
GenBank, https://identifiers.org/insdc:GHIW00000000 (2018).
GenBank, https://identifiers.org/insdc:GHIX00000000 (2018).
NGSPipeline interface, http://ngspipelines.toulouse.inra.fr:9014 (2017)
Péden R. et al. Environmental transcriptomes of invasive dreissena: a move towards an upcoming model species. figshare, https://doi.org/10.6084/m9.figshare.c.4483856 (2019).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome res. 12, 656–664 (2002).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Acknowledgements
This work was supported by the the BIOMICS program (Biomarkers and Omics in the freshwater Bivalve Dreissena polymorpha) of the French Agency for Biodiversity/National Agency for Water and Aquatic Environments (AFB/ONEMA) Authors would like to thanks the GeT-PlaGe for performing sequencing and Genotoul platform for transcriptome assembly, annotation and validation.
Author information
Authors and Affiliations
Contributions
B.S., S.D., P.P., L.G. and S.P.-D. conceived and designed the study. B.S., J.F., S.P.-D., M.P. and F.L. realized experiments. C.K. and R.-F.S. performed sequencing and transcriptome assembly and annotation. B.S., R.P. and P.P. proceeded to data analysis. R.P., B.S., S.D., R.-F.S. and C.K. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Péden, R., Poupin, P., Sohm, B. et al. Environmental transcriptomes of invasive dreissena, a model species in ecotoxicology and invasion biology. Sci Data 6, 234 (2019). https://doi.org/10.1038/s41597-019-0252-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0252-x
This article is cited by
-
Mechanisms underlying Clostridium pasteurianum’s metabolic shift when grown with Geobacter sulfurreducens
Applied Microbiology and Biotechnology (2022)
