
Abstract
The normally soluble TAR DNA-binding protein 43 (TDP-43) is found aggregated both in reversible stress granules and in irreversible pathogenic amyloid. In TDP-43, the low-complexity domain (LCD) is believed to be involved in both types of aggregation. To uncover the structural origins of these two modes of β-sheet-rich aggregation, we have determined ten structures of segments of the LCD of human TDP-43. Six of these segments form steric zippers characteristic of the spines of pathogenic amyloid fibrils; four others form LARKS, the labile amyloid-like interactions characteristic of protein hydrogels and proteins found in membraneless organelles, including stress granules. Supporting a hypothetical pathway from reversible to irreversible amyloid aggregation, we found that familial ALS variants of TDP-43 convert LARKS to irreversible aggregates. Our structures suggest how TDP-43 adopts both reversible and irreversible β-sheet aggregates and the role of mutation in the possible transition of reversible to irreversible pathogenic aggregation.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Change history
17 September 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Kato, M. et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012).
Conicella, A. E., Zerze, G. H., Mittal, J. & Fawzi, N. L. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549 (2016).
Kato, M. & McKnight, S. L. A solid-state conceptualization of information transfer from gene to message to protein. Annu. Rev. Biochem. https://doi.org/10.1146/annurev-biochem-061516-044700 (2017).
Marcotte, E. M., Pellegrini, M., Yeates, T. O. & Eisenberg, D. A census of protein repeats. J. Mol. Biol. 293, 151–160 (1999).
Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 (2005).
Zoghbi, H. Y. & Orr, H. T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23, 217–247 (2000).
March, Z. M., King, O. D. & Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 1647, 9–18 (2016).
Bentmann, E. et al. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094 (2012).
Colombrita, C. et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061 (2009).
Dewey, C. M. et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31, 1098–1108 (2011).
Gilks, N. et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398 (2004).
Dang, Y. et al. Eukaryotic initiation factor 2α-independent pathway of stress granule induction by the natural product pateamine A. J. Biol. Chem. 281, 32870–32878 (2006).
Kedersha, N. et al. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151, 1257–1268 (2000).
Patel, A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015).
Reineke, L. C. et al. Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 37, e00596–e16 (2017).
Murray, D. T. et al. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615–627.e16 (2017).
Wheeler, J. R., Matheny, T., Jain, S., Abrisch, R. & Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 5, 1–25 (2016).
Jain, A. & Vale, R. D. RNA phase transitions in repeat expansion disorders. Nature 546, 243–247 (2017).
Lin, Y., Protter, D. S. W., Rosen, M. K. & Parker, R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015).
Hughes, M. P. et al. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701 (2018).
Eisenberg, D. S. & Sawaya, M. R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95 (2017).
Knowles, T. P. J., Vendruscolo, M. & Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014).
Meersman, F. & Dobson, C. M. Probing the pressure-temperature stability of amyloid fibrils provides new insights into their molecular properties. Biochim. Biophys. Acta. 1764, 452–460 (2006).
Harrison, A. F. & Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474, 1417–1438 (2017).
Chang, C. K. et al. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 425, 219–224 (2012).
Lukavsky, P. J. et al. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449 (2013).
Saini, A. & Chauhan, V. S. Self-assembling properties of peptides derived from TDP-43 C-terminal fragment. Langmuir 30, 3845–3856 (2014).
Buratti, E. & Baralle, F. E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429 (2010).
Lee, E. B., Lee, V. M. Y. & Trojanowski, J. Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50 (2011).
Schwab, C., Arai, T., Hasegawa, M., Yu, S. & McGeer, P. L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 67, 1159–1165 (2008).
Amador-Ortiz, C. et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435–445 (2007).
Nakashima-Yasuda, H. et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229 (2007).
Higashi, S. et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 1184, 284–294 (2007).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Kabashi, E. et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 (2008).
Chiang, C.-H. et al. Structural analysis of disease-related TDP-43 D169G mutation: linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci. Rep. 6, 21581 (2016).
Lagier-Tourenne, C., Polymenidou, M. & Cleveland, D. W. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19R1, R46–R64 (2010).
Pesiridis, G. S., Lee, V. M. Y. & Trojanowski, J. Q. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 18R1, R156–R162 (2009).
Igaz, L. M. et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 284, 8516–8524 (2009).
Hasegawa, M. et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 (2008).
Ling, S. C., Polymenidou, M. & Cleveland, D. W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013).
Ling, J. P., Pletnikova, O., Troncoso, J. C. & Wong, P. C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015).
Yang, C. et al. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 5, e15878 (2010).
Chen, A. K. et al. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187 (2010).
Cushman, M., Johnson, B. S., King, O. D., Gitler, A. D. & Shorter, J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J. Cell Sci. 123, 1191–1201 (2010).
Guo, W. et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830 (2011).
Jiang, L. L. et al. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288, 19614–19624 (2013).
Jiang, L. L. et al. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 6, 23928 (2016).
Mompeán, M. et al. “Structural characterization of the minimal segment of TDP-43 competent for aggregation”. Arch. Biochem. Biophys. 545, 53–62 (2014).
Sawaya, M. R. et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447, 453–457 (2007).
Goldschmidt, L., Teng, P. K., Riek, R. & Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 107, 3487–3492 (2010).
Budini, M. et al. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287, 7512–7525 (2012).
Fuentealba, R. A. et al. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 285, 26304–26314 (2010).
Seidler, P. M. et al. Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176 (2018).
Xiang, S. et al. The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell 163, 829–839 (2015).
Gitcho, M. A. et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538 (2008).
Xu, M. et al. Characterization of β-domains in C-terminal fragments of TDP-43 by scanning tunneling microscopy. J. Struct. Biol. 181, 11–16 (2013).
Lim, L., Wei, Y., Lu, Y. & Song, J. ALS-Causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 14, e1002338 (2016).
Estes, P. S. et al. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321 (2011).
Walker, A. K. et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One 8, e81170 (2013).
Bosco, D. A. et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175 (2010).
Hackman, P. et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann. Neurol. 73, 500–509 (2013).
Kim, H. J. et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013).
Schmidt, H. B. & Rohatgi, R. In vivo formation of vacuolated multi-phase compartments lacking membranes. Cell Reports 16, 1228–1236 (2016).
Franzmann, T. M. et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 359, eaao5654 (2018).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Soragni, A. et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 (2016).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Cheng, P.-N., Liu, C., Zhao, M., Eisenberg, D. & Nowick, J. S. Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 4, 927–933 (2012).
Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Wiltzius, J. J. et al. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin). Protein Sci. 17, 1467–1474 (2008).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 (1993).
Rodriguez, J. A. et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 (2015).
Hattne, J. et al. MicroED data collection and processing. Acta Crystallogr. A Found. Adv. 71, 353–360 (2015).
Shi, D. et al. The collection of MicroED data for macromolecular crystallography. Nat. Protoc. 11, 895–904 (2016).
Bricogne, G. et al. BUSTER v.2.9. (Global Phasing Ltd., Cambridge, 2010).
Lawrence, M. C. & Colman, P. M. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 (1993).
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
Acknowledgements
We thank S. Sangwan and P. Seidler for discussion, D. Shi and T. Gonen at Janelia for microscope support and M. Collazo at UCLA-DOE Macromolecular Crystallization Core Technology Center for crystallization support. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Eiger 16 M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research was supported in part by grants from the National Institutes of Health NIH NIA AG029430, NIH NIA AG054022, Howard Hughes Medical Institute and the Janelia Research Campus visitor program. This material is based upon work supported by the National Science Foundation under Grant No. NSF 1616265. We acknowledge the use of instruments at the Electron Imaging Center for Nanomachines supported by UCLA and by instrumentation grants from NIH (1S10RR23057 and 1U24GM116792) and NSF (DBI-1338135). D.R.B. was supported by the National Science Foundation Graduate Research Fellowship Program.
Author information
Authors and Affiliations
Contributions
E.L.G., Q.C. and D.S.E. designed the project and wrote the manuscript with input from all other authors, especially M.R.S. E.L.G. and H.T. conducted fibril growth assays and prepared peptide crystals. E.L.G., Q.C. and J.L. cloned and purified TDP-43 constructs and performed the protein aggregation assay and fibril diffraction. M.P.H. predicted putative LARKS. E.L.G., H.T. and M.R.S. processed and solved 300GNNQGSN306. E.L.G. and M.R.S. processed and solved 312NFGAFS317, 312NFGTFS317, 370GNNSYS375 and 396GFNGGFG402. E.L.G., Q.C., J.L. and M.R.S. processed data and solved the 321AMMAAA326 and 328AALQSS333. J.A.R. collected MicroED data on 312NFGEFS317 and 333SWGMMGMLASQ343, M.R.S., D.C. and E.L.G. processed data and solved the structure of 312NFGEFS317 and 333SWGMMGMLASQ343. D.R.B. collected MicroED data for 312NFGpTFS317. Q.C., M.R.S., D.C. and D.R.B. processed data and solved the structure of 312NFGpTFS317. E.L.G., Q.C., M.R.S., M.P.H. and D.S.E. analyzed structures and designed the model of the LCD in SG formation and pathological aggregation.
Corresponding author
Ethics declarations
Competing interests
D.S.E. is an advisor and equity shareholder in ADDRx, Inc.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated Supplementary Information
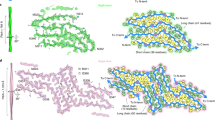
Supplementary Figure 1 321AMMAAA326 and 333SWGMMGMLASQ343 form antiparallel steric-zipper structures.
Views perpendicular to the fibril axis (vertical) are shown for three steric-zipper structures of Fig. 1. 321AMMAAA326 and 333SWGMMGMLASQ343 are two zippers that adopt anti-parallel packing, as shown by the alternating orientation of arrows representing the direction of strands. 328AALOSS333 is shown as an example of the zippers which adopt parallel packing, as shown by the same orientation of arrows in each sheet.
Supplementary Figure 2 Validation of TDP-CTF and TDP-LCD constructs.
The SUMO tagged TDP-CTF and TDP-LCD were analyzed by SDS-PAGE and stained with coomassie blue and anti-TDP-43 antibody. For TDP-CTF, notice that 1 hour after (1h) ULP1 cleavage, it generates SUMO protein and tag-free TDP-CTF, and both SUMO-tagged and tag-free TDP-CTF can be recognized by anti-TDP-43 antibody. SUMO tagged TDP-LCD also can be recognized by anti-TDP-43 antibody, and generates SUMO protein and tag-free TDP-LCD after cleavage. The molecular weight of SUMO and tag-free TDP-LCD are so close to each other that the two bands are indistinguishable on SDS-PAGE when stained by coomassie blue. However, the existence of tag-free TDP-LCD can be detected by anti-TDP-43 antibody, forming a band (although very dim) at its expected position. Tag-free TDP-LCD can also be detected by SDS-PAGE of the TDP-LCD pellet, showing a band at its expected position, and SUMO protein remains soluble as shown in Fig. 2a.
Supplementary Figure 3 Positive control of denaturing conditions shown in Fig. 3b and structural details of 312NFGEFS317 and 312NFGpTFS317.
(A) Negative stain EM images of Aβ1-42 fibrils, typical amyloid fibrils, before and after treatment with 2% SDS and 70 °C for 15 mins. Notice that Aβ1-42 fibrils are stable and maintain their morphology after treatment in denaturing conditions. Scale bar = 200 nm. (B) Omit map of phosphate in 312NFGpTFS317 structure viewed from two angles. Green mesh shows the electron density of the Fo-Fc map with sigma > 4.5. Notice that the electron density of the same map with sigma <-4.5 is also computed as red mesh, however there is no observable negative density. TPO: phosphorylated threonine. (C) Hydrogen bond networks that stabilizes the packing of 312NFGEFS317 and 312NFGpTFS317. In each structure, three Glu/pThr residues from three adjacent strands of same sheet are shown, as well as the residues hydrogen-bonded to them. The hydrogen bonds formed by all Glu and the central pThr are shown as yellow dash lines, with distances between 2.3 and 3.2 Å.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3 and Supplementary Tables 1 and 2
Rights and permissions
About this article

Cite this article
Guenther, E.L., Cao, Q., Trinh, H. et al. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat Struct Mol Biol 25, 463–471 (2018). https://doi.org/10.1038/s41594-018-0064-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-018-0064-2
This article is cited by
-
Halogen doped graphene quantum dots modulate TDP-43 phase separation and aggregation in the nucleus
Nature Communications (2024)
-
Cryo-EM observation of the amyloid key structure of polymorphic TDP-43 amyloid fibrils
Nature Communications (2024)
-
Advances in mass spectrometry to unravel the structure and function of protein condensates
Nature Protocols (2023)
-
Targeting the glycine-rich domain of TDP-43 with antibodies prevents its aggregation in vitro and reduces neurofilament levels in vivo
Acta Neuropathologica Communications (2023)
-
Low complexity domains of the nucleocapsid protein of SARS-CoV-2 form amyloid fibrils
Nature Communications (2023)
