
Abstract
Critical COVID-19 is caused by immune-mediated inflammatory lung injury. Host genetic variation influences the development of illness requiring critical care1 or hospitalization2,3,4 after infection with SARS-CoV-2. The GenOMICC (Genetics of Mortality in Critical Care) study enables the comparison of genomes from individuals who are critically ill with those of population controls to find underlying disease mechanisms. Here we use whole-genome sequencing in 7,491 critically ill individuals compared with 48,400 controls to discover and replicate 23 independent variants that significantly predispose to critical COVID-19. We identify 16 new independent associations, including variants within genes that are involved in interferon signalling (IL10RB and PLSCR1), leucocyte differentiation (BCL11A) and blood-type antigen secretor status (FUT2). Using transcriptome-wide association and colocalization to infer the effect of gene expression on disease severity, we find evidence that implicates multiple genes—including reduced expression of a membrane flippase (ATP11A), and increased expression of a mucin (MUC1)—in critical disease. Mendelian randomization provides evidence in support of causal roles for myeloid cell adhesion molecules (SELE, ICAM5 and CD209) and the coagulation factor F8, all of which are potentially druggable targets. Our results are broadly consistent with a multi-component model of COVID-19 pathophysiology, in which at least two distinct mechanisms can predispose to life-threatening disease: failure to control viral replication; or an enhanced tendency towards pulmonary inflammation and intravascular coagulation. We show that comparison between cases of critical illness and population controls is highly efficient for the detection of therapeutically relevant mechanisms of disease.
Similar content being viewed by others


Main
Critical illness in COVID-19 is both an extreme disease phenotype and a relatively homogeneous clinical definition; it includes patients with hypoxaemic respiratory failure5 with acute lung injury6, and excludes many patients with non-pulmonary clinical presentations7, who are known to have divergent responses to therapy8. In the UK, individuals in the critically ill group are younger, less likely to have significant comorbidity and more severely affected than a general hospitalized cohort5, characteristics which may amplify observed genetic effects. In addition, as development of critical illness is in itself a key clinical end-point for therapeutic trials8, using critical illness as a phenotype in genetic studies enables the detection of directly therapeutically relevant genetic effects1.
Using microarray genotyping in 2,244 cases, we previously discovered that critical COVID-19 is associated with genetic variation in the host immune response to viral infection (OAS1, IFNAR2 and TYK2) and the inflammasome regulator DPP91. In collaboration with international groups, we extended these findings to include a variant near TAC4 (rs77534576)3. Several variants have been associated with milder phenotypes, including the ABO blood-type locus2, a pleiotropic inversion in chr17q21.319 and associations in five additional loci, including the T lymphocyte-associated transcription factor, FOXP43. An enrichment of rare loss-of-function variants in candidate interferon signalling genes has been reported4, but this has yet to be replicated at genome-wide significance thresholds10,11.
In partnership with Genomics England, we performed whole-genome sequencing (WGS) to improve the resolution and deepen the fine-mapping of significant signals and thereby provide further biological insight into critical COVID-19. Here we present results from a cohort of 7,491 critically ill patients from 224 intensive care units, compared with 48,400 control individuals, describing the discovery and validation of 23 gene loci for susceptibility to critical COVID-19 (Extended Data Fig. 1).
Genome-wide association study analysis
After quality control procedures, we used a logistic mixed model regression, implemented in SAIGE12, to perform association analyses with unrelated individuals (critically ill cases, n = 7,491; controls, n = 48,400 (100,000 Genomes Project (100k) cohort, n = 46,770; mild COVID-19, n = 1,630) (Methods, Supplementary Table 2). A total of 1,339 of these cases were included in the primary analysis for our previous report1. Genome-wide association studies (GWASs) were performed separately for genetic ancestry groups (ncases/ncontrols: European (EUR) 5,989/42,891; South Asian (SAS) 788/3,793; African (AFR) 440/1,350; East Asian (EAS) 274/366), and combined by inverse-variance-weighted fixed effects meta-analysis using METAL (Methods). We established the independence of signals using GCTA-cojo, and we validated this with conditional analysis using individual-level data with SAIGE (Methods, Supplementary Table 6). To reduce the risk of spurious associations arising from genotyping or pipeline errors, we required supporting evidence from variants in linkage disequilibrium (LD) for all genome-wide-significant variants: observed z-scores for each variant were compared with imputed z-scores for the same variant, with discrepant values being excluded (see Methods, Supplementary Fig. 2).
In population-specific analyses, we discovered 22 independent genome-wide-significant associations in the EUR ancestry group (Fig. 1, Supplementary Fig. 11, Table 1) at a \(P\) value threshold adjusted for multiple testing (2.2 × 10−08; Supplementary Table 5). In multi-ancestry meta-analysis, we identified an additional three independent genome-wide-significant association signals (Fig. 1, Table 1).

Manhattan plots are shown on the left and quantile–quantile (QQ) plots of observed versus expected \(P\) values on the right, with genomic inflation (λ) displayed for each analysis. Highlighted results in blue in the Manhattan plots indicate variants that are LD-clumped (r2 = 0.1, P2 = 0.01, EUR LD) with the lead variants at each locus. Gene name annotation indicates genes that are affected by the predicted worst consequence type of each lead variant (annotation by Variant Effect Predictor (VEP)). For the HLA locus, the gene that was identified by HLA allele analysis is annotated. The GWAS was performed using logistic regression and meta-analysed by the inverse variant method. The red dashed line shows the Bonferroni-corrected P value: P = 2.2 × 10−8.
To assess the sensitivity of our results to mismatches of demographic characteristics between cases and controls (Supplementary Figs. 9, 10), we performed an age-, sex- and body mass index (BMI)-matched case–control analysis (Supplementary Figs. 18–21). As there is a theoretical risk of mismatch between cases and 100,000 Genomes Project participants in risk factors for exposure (for example, shielding behaviour) or susceptibility to critical COVID-19 (for example, immunosuppression), we performed a sensitivity analysis using only the cohort with mild COVID-19 (see above; Supplementary Table 10). In both of these analyses, allele frequencies and directions of effect were concordant for all lead signals.
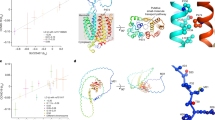
We inferred credible sets of variants using Bayesian fine-mapping with susieR13, by analysing the GWAS summaries of 17 regions of genomic length 3 Mb that were flanking groups of lead signals. We obtained 22 independent credible sets of variants for EUR and an additional 2 from the trans-ancestry meta-analysis with a posterior inclusion probability greater than 0.95 (Extended Data Table 1, Supplementary Information). Fine-mapping of the association signals revealed putative causal variants for both previously reported and novel association signals (see Supplementary Information, Extended Data Table 1). In 12 out of the 24 fine-mapped signals, the credible sets included 5 or fewer variants, and for 8 signals we detected variants with predicted missense or worse consequence across each credible set (Extended Data Table 1). We were able to fine-map multiple independent signals at previously identified loci (Fig. 3, Extended Data Figs. 2, 4). For example, the signal in the 3p21.31 region2, was fine-mapped into two independent associations, with the credible set for the first refined to a single variant in the 5′ untranslated region (UTR) of SLC6A20 (chr3:45796521:G:T, rs2271616, odds ratio (OR): 1.29, 95% confidence interval (CI):1.21–1.37), and the second credible set including multiple variants in downstream and intronic regions of LZTFL1 (Fig. 3). Among the novel signals, at 3q24 and 9p21.3 we detected missense variants that affect PLSCR1 and IFNA10, respectively (chr3:146517122:G:A, rs343320, p.His262Tyr, OR: 1.24, 95% CI: 1.15–1.33, CADD: 22.6; chr9:21206606:C:G, rs28368148, p.Trp164Cys, OR:1.74, 95% CI: 1.45–2.09, CADD: 23.9). Both are predicted to be deleterious by the Combined Annotation Dependent Depletion (CADD) tool14. Structural predictions for these variants suggest functional effects (Extended Data Fig. 5). We assessed whether the main signals of this study were underlain by rarer variants with a lower minor allele frequency (MAF) (less than 0.02%) than our GWAS default threshold (less than 0.5%), by including rarer variant summaries when fine-mapping, but no additional variants were added to the main credible sets (Supplementary Table 9).
Consistent with our expectation that genetic susceptibility has a stronger role in younger individuals, age-stratified analysis (individuals of younger than 60 years old versus individuals of 60 years old or above) in the EUR group revealed a signal in the 3p21.31 region with a significantly stronger effect in the younger age group (chr3:45801750:G:A, rs13071258, OR: 3.34, 95% CI: 2.98–3.75 versus OR: 2.1, 95% CI 1.88–2.34), which is in strong LD (r2 = 0.947) with the main GWAS signal indexed by rs73064425. Sex-specific analysis did not reveal significant effects (Supplementary Fig. 17).
Replication
For replication, we performed a meta-analysis of summary statistics generously shared by 23andMe and the COVID-19 Host Genetics Initiative (HGI) data freeze 6 (B2). As a previous analysis of GenOMICC1 contributes a substantial part of the signal at each locus in HGI v.6, and leave-one-out analyses were not available, we removed the signal from GenOMICC cases in HGI v.6 using mathematical subtraction to ensure independence (Methods). Using LD clumping to find variants genotyped in both the discovery and replication studies, we required P < 0.002 (0.05/25) and concordant direction of effect (Table 1, Supplementary Table 8) for replication. We interrogated two variants that failed replication in this set in a second GWAS meta-analysis of hospitalized patients with COVID-19 from UK Biobank, AncestryDNA, Penn Medicine Biobank and Geisinger Health Systems, which included a total of 9,937 individuals who were hospitalized with COVID-19 and 1,059,390 control individuals. This led to a further successful replicated finding, in IFNA10 (Table 1).
We replicated 23 of the 25 significant associations that were identified in the population-specific and/or multi-ancestry GWASs. One of the non-replicated signals (rs4424872) corresponds to a rare variant that may not be well represented in the replication datasets—which are dominated by single-nucleotide polymorphism (SNP) genotyping data—but which also had significant heterogeneity among ancestries. The second non-replicated signal is within the human leukocyte antigen (HLA) locus, which has complex LD (see below).
HLA region
The lead variant in the HLA region, rs9271609, lies upstream of the HLA-DQA1 and HLA-DRB1 genes. To investigate the contribution of specific HLA alleles to the observed association in the HLA region, we imputed HLA alleles at a four-digit (two-field) level using HIBAG15. The only allele that reached genome-wide significance was HLA-DRB1*04:01 (OR: 0.80, 95% CI: 0.75–0.86, P = 1.6 × 10−10 in EUR), which has a stronger \(P\) value than the lead SNP in the region (OR: 0.88, 95% CI: 0.84–0.92, P = 3.3 × 10−9 in EUR) and is a better fit to the data (Akaike information criterion (AIC): AICDRB1*04:01 = 30,241.34; AICleadSNP = 30,252.93) (Extended Data Fig. 6). HLA-DRB1*04:01 has been previously reported to confer protection against severe disease in a small cohort of European ancestry16.
Gene burden testing
To assess the contribution of rare variants to critical illness, we performed gene-based analysis using SKAT-O as implemented in SAIGE-GENE17 on a subset of 12,982 individuals from our cohort (7,491 individuals with critical COVID-19 and 5,391 control individuals), for which the genome-sequencing data were processed with the same alignment and variant calling pipeline. We tested the burden of rare (MAF < 0.5%) variants considering the predicted variant consequence type (tested variant counts provided in the Supplementary Information). We assessed burden using a strict definition for damaging variants (high-confidence putative loss-of-function (pLoF) variants as identified by LOFTEE18) and a lenient definition (pLoF plus missense variants with CADD ≥ 10)14, but found no significant associations at a gene-wide-significance level. Moreover, all individual rare variants included in the tests had P values greater than 10−5.
Consistent with other recent work11, we did not find any significant gene burden test associations among the 13 genes previously reported from an interferon-pathway-focused study4 (tests for all genes had P > 0.05; Supplementary Information), and we did not replicate the reported association19,20,21 in TLR7 (EUR P = 0.30 for pLoF and P = 0.075 for missense variants).
Transcriptome-wide association study analysis
To infer the effect of genetically determined variation in gene expression on disease susceptibility, we performed a transcriptome-wide association study (TWAS) using gene expression data (GTEx v.8; ref. 22) for two disease-relevant tissues: lung and whole blood. We found significant associations between critical COVID-19 and predicted expression in lung (14 genes) and blood (6 genes) (Supplementary Fig. 23) and in an all-tissue meta-analysis (GTEx v.8; 51 genes) (Supplementary Fig. 24). Expression signals for 16 genes significantly colocalized with susceptibility (Fig. 2). As the LD structure of the HLA is complex, we only assessed colocalization for the significant association, HLA-DRB1. Although it was not significant in our TWAS analysis, expression quantitative trait loci (eQTLs) in the proximity of the association significantly colocalize with the GWAS signal for both blood and lung (both PPH4 > 0.8; Supplementary Information).

The highlighting colour is different for the lung and blood tissue data that were used for colocalization, and we also distinguish loci that were significant in both. Results are grouped according to two classes for the posterior probability of colocalization (PPH4): P > 0.5 and P > 0.8. If a variant is placed in both classes, then the colour that corresponds to the higher probability class is shown. Arrowheads indicate the direction of change in gene expression associated with an increased disease risk. The red dashed line shows the Bonferroni-corrected significance threshold for the metaTWAS analysis at P = 2.3 × 10−6.
We repeated the TWAS analysis using models of intron excision rate from GTEx v.8 to obtain a splicing TWAS, which revealed significant signals in lung (16 genes) and whole blood (9 genes), and in an all-tissue meta-analysis (33 genes); 11 of these had strongly colocalizing splicing signals (Supplementary Information).
Mendelian randomization
We performed generalized summary-data-based Mendelian randomization (GSMR)23 in a replicated outcome study design using the protein quantitative trait loci (pQTLs) from the INTERVAL study24. GSMR incorporates information from multiple independent SNPs and provides stronger evidence of a causal relationship than single-SNP-based approaches. Of 16 proteome-wide-significant associations in this study, 8 were replicated in an external dataset at a Bonferroni-corrected P value threshold of P < 0.0031 (P < 0.05/16; Extended Data Table 2, Extended Data Fig. 7) .

Top two panels, variants in LD with the lead variants shown. The variants that are included in two independent credible sets are displayed with black outline circles. The r2 values in the key denote upper limits; that is, 0.2 = [0, 0.2], 0.4 = [0.2, 0.4], 0.6 = [0.4, 0.6], 0.8 = [0.6, 0.8],1 = [0.8, 1]. Bottom, locations of protein-coding genes, coloured by TWAS P value. The red dashed line shows the Bonferroni-corrected P value: P = 2.2 × 10−8 for individuals of European ancestry.
Discussion
We report 23 replicated genetic associations with critical COVID-19, which were discovered in only 7,491 cases. This demonstrates the efficiency of the design of the GenOMICC study, an open-source25 international research programme (https://genomicc.org) that focuses on extreme phenotypes: patients with life-threatening infectious disease, sepsis, pancreatitis and other critical illness phenotypes. GenOMICC detects greater heritability and stronger effect sizes than other study designs across all variants (Supplementary Figs. 22, 14). In COVID-19, critical illness is not only an extreme susceptibility phenotype, but also a more homogeneous one: we have shown previously that critically ill patients with COVID-19 are more likely to have the primary disease process—hypoxaemic respiratory failure5—and that patients in this group have a divergent response to immunosuppressive therapy compared to other hospitalized patients8. We detect distinct signals at several of the associated loci, in some cases implicating different biological mechanisms.
Five of the variants associated with critical COVID-19 have direct roles in interferon signalling and broadly concordant predicted biological effects. These include a probable destabilizing amino acid substitution in a ligand, IFNA10 (Trp164Cys, Extended Data Fig. 5), and—as we reported previously1—reduced expression of a subunit of its receptor IFNAR2 (Fig. 2). IFNAR2 signals through a kinase that is encoded by TYK21. Although the lead variant in TYK2 in WGS is a protein-coding variant with reduced STAT1 phosphorylation activity26, it is also associated with significantly increased expression of TYK2 (Fig. 2, Methods). Fine-mapping reveals a significant association with an independent missense variant in IL10RB, a receptor for type III (lambda) interferons (rs8178521; Table 1). Finally, we detected a lead risk variant in phospholipid scramblase 1 (chr3:146517122:G:A, rs343320; PLSCR1) which disrupts a nuclear localization signal that is important for the antiviral effect of interferons27 (Extended Data Fig. 5). PLSCR1 controls the replication of other RNA viruses, including vesicular stomatitis virus, encephalomyocarditis virus and influenza A virus27,28.
Although our genome-wide gene-based association tests did not replicate any findings from a previous pathway-specific study of rare deleterious variants4, our results provide robust evidence implicating reduced interferon signalling in susceptibility to critical COVID-19. Notably, systemic administration of interferon in two large clinical trials, albeit late in disease, did not reduce mortality29,30.
We found significant associations in genes that are implicated in lymphopoesis and in the differentiation of myeloid cells. BCL11A is essential for B and T lymphopoiesis31 and promotes the differentiation of plasmacytoid dendritic cells32. TAC4, reported previously3, encodes a regulator of B cell lymphopoesis33 and antibody production34, and promotes the survival of dendritic cells35. Finally, although the strongest fine-mapping signal at 5q31.1 (chr5:131995059:C:T, rs56162149) is in an intron of ACSL6 with significant effects on expression (Supplementary Information), the credible set includes a missense variant in CSF2 (encoding granulocyte–macrophage colony stimulating factor; GM-CSF) of uncertain significance (chr5:132075767:T:C; Extended Data Table 1). We have previously shown that GM-CSF is strongly up-regulated in critical COVID-1936, and it is already under investigation as a target for therapy37. Mendelian randomization results are consistent with a direct link between the plasma levels of a closely related cytokine receptor subunit, IL3RA, and critical COVID-19 (Extended Data Table 2).
Fine-mapping, colocalization and TWAS analyses provide evidence for increased expression of MUC1 as the mediator of the association with rs41264915 (Supplementary Table 12). This suggests that mucins could have a therapeutically important role in the development of critical illness in COVID-19.
Mendelian randomization provides genetic evidence in support of a causal role for coagulation factors (F8) and platelet activation (PDGFRL) in critical COVID-19 (Extended Data Table 2, Extended Data Fig. 7), consistent with autopsy6, proteomic38 and therapeutic39 evidence. Perhaps more importantly, we identify specific and closely related intercellular adhesion molecules that have known roles in the recruitment of inflammatory cells to sites of inflammation, including E-selectin (SELE), intercellular adhesion molecule 5 (ICAM5) and DC-SIGN (dendritic-cell-specific ICAM3-grabbing non-integrin; CD209), which may provide additional therapeutic targets. DC-SIGN (CD209) mediates pathogen endocytosis and antigen presentation, and is known to be involved in multiple viral infections, including SARS-CoV and influenza A virus. It has affinity for SARS-CoV-240,41.
Our previous report of an association between the OAS gene cluster and severe disease was robustly replicated in an external cohort1, but does not meet genome-wide significance in the present analysis (Supplementary Table 7). This may indicate a change in the observed effect size because any effect that is detected in GWASs is more likely to have been sampled from the larger end of the range of possible effect sizes —the ‘winner’s curse’. Alternatively, it may indicate either a change in the population of patients (cases or controls) or a change in the pathogen. For example it is possible that—as with the other coronaviruses that are known to infect humans42—more recent variants of SARS-CoV-2 have evolved to overcome this host antiviral defence mechanism.
Limitations
In contrast to microarray genotyping, WGS is a rapidly evolving and relatively new technology for GWASs, with relatively few sources of population controls. We selected a control cohort from the 100,000 Genomes Project, which was sequenced and analysed using a different platform and bioinformatics pipeline compared with the case cohort (Extended Data Fig. 1). However, to minimize the risk of false-positive associations due to technical artifacts, extensive quality measures were used (Methods). In brief, we masked low-quality genotypes, filtered for genotype signal using a low threshold for missingness and performed a control–control relative allele frequency filter using a subset of samples processed with both bioinformatics pipelines. Finally, we required all significant associations to be supported by local variants in LD, which may be excessively stringent (Methods). Although this approach may remove some true associations, our priority is to maximize confidence in the reported signals. Of 25 variants that meet this requirement, 23 are externally replicated, and the remaining 2 may be true associations that are yet to be replicated owing to a lack of coverage or power in the replication datasets.
The design of our study incorporates genetic signals for every stage in the disease progression into a single phenotype. This includes establishment of infection, viral replication, inflammatory lung injury and hypoxaemic respiratory failure. Although we can have considerable confidence that the replicated associations with critical COVID-19 we report are robust, we cannot determine at which stage in the disease process, or in which tissue, the relevant biological mechanisms are active.
Conclusions
These genetic associations identify biological mechanisms that may underlie the development of life-threatening COVID-19, several of which may be amenable to therapeutic targeting. Furthermore, we demonstrate the value of WGS for fine-mapping loci in a complex trait. In the context of the ongoing global pandemic, translation to clinical practice is an urgent priority. As with our previous work, biological and molecular studies—and, where appropriate, large-scale randomized trials—will be essential before our findings can be translated into clinical practice.
Methods
Ethics
GenOMICC study: GenOMICC was approved by the following research ethics committees: Scotland ‘A’ Research Ethics Committee (15/SS/0110) and Coventry and Warwickshire Research Ethics Committee (England, Wales and Northern Ireland) (19/WM/0247). Current and previous versions of the study protocol are available at https://genomicc.org/protocol/. 100,000 Genomes project: the 100,000 Genomes project was approved by the East of England—Cambridge Central Research Ethics Committee (REF 20/EE/0035). Only individuals from the 100,000 Genomes project for whom WGS data were available and who consented for their data to be used for research purposes were included in the analyses. UK Biobank study: ethical approval for the UK Biobank was previously obtained from the North West Centre for Research Ethics Committee (11/NW/0382). The work described herein was approved by UK Biobank under application number 26041. Geisinger Health Systems (GHS) study: approval for DiscovEHR analyses was provided by the GHS Institutional Review Board under project number 2006-0258. AncestryDNA study: all data for this research project were from individuals who provided prior informed consent to participate in AncestryDNA’s Human Diversity Project, as reviewed and approved by our external institutional review board, Advarra (formerly Quorum). All data were de-identified before use. Penn Medicine Biobank study: appropriate consent was obtained from each participant regarding the storage of biological specimens, genetic sequencing and genotyping, and access to all available EHR data. This study was approved by the institutional review board of the University of Pennsylvania and complied with the principles set out in the Declaration of Helsinki. Informed consent was obtained for all study participants. 23andMe study: participants in this study were recruited from the customer base of 23andMe, a personal genetics company. All individuals included in the analyses provided informed consent and answered surveys online according to the 23andMe protocol for research in humans, which was reviewed and approved by Ethical and Independent Review Services, a private institutional review board (http://www.eandireview.com).
Recruitment of cases (patients with COVID-19)
Patients were recruited to the GenOMICC study in 224 UK intensive care units (https://genomicc.org). All individuals had confirmed COVID-19 according to local clinical testing and were deemed, in the view of the treating clinician, to require continuous cardiorespiratory monitoring. In UK practice this kind of monitoring is undertaken in high-dependency or intensive care units.
Recruitment of control individuals
Mild or asymptomatic control individuals
Participants were recruited to the mild COVID-19 cohort on the basis of having experienced mild (non-hospitalized) or asymptomatic COVID-19. Participants volunteered to take part in the study via a microsite and were required to self-report the details of a positive COVID-19 test. Volunteers were prioritized for genome sequencing on the basis of demographic matching with the critical COVID-19 cohort considering self-reported ancestry, sex, age and location within the UK. We refer to this cohort as the COVID-19 mild cohort.
Control individuals from the 100,000 Genomes project
Participants were enrolled in the 100,000 Genomes Project from families with a broad range of rare diseases, cancers and infection by 13 regional NHS Genomic Medicine Centres across England and in Northern Ireland, Scotland and Wales. For this analysis, participants for whom a positive SARS-CoV-2 test had been recorded as of March 2021 were not included owing to uncertainty in the severity of COVID-19 symptoms. Only participants for whom genome sequencing was performed from blood-derived DNA were included and participants with haematological malignancies were excluded to avoid potential tumour contamination.
DNA extraction
For severe cases of COVID-19 and mild cohort controls, DNA was extracted from whole blood either manually using a Nucleon Kit (Cytiva) and resuspended in 1 ml TE buffer pH 7.5 (10 mM Tris-Cl pH 7.5, 1 mM EDTA pH 8.0), or automated on the Chemagic 360 platform using the Chemagic DNA blood kit (PerkinElmer) and re-suspended in 400 μl elution buffer. The yield of the DNA was measured using Qubit and normalized to 50 ng μl−1 before sequencing. For the 100,000 Genomes Project samples, DNA was extracted from whole blood at designated extraction centres following sample handling guidance provided by Genomics England and NHS England.
WGS
Sequencing libraries were generated using the Illumina TruSeq DNA PCR-Free High Throughput Sample Preparation kit and sequenced with 150-bp paired-end reads in a single lane of an Illumina Hiseq X instrument (for 100,000 Genomes Project samples) or a NovaSeq instrument (for the COVID-19 critical and mild cohorts).
Sequencing data quality control
All genome sequencing data were required to meet minimum quality metrics and quality control measures were applied for all genomes as part of the bioinformatics pipeline. The minimum data requirements for all genomes were: more than 85 × 10−9 bases with Q ≥ 30 and at least 95% of the autosomal genome covered at 15× or higher calculated from reads with mapping quality greater than 10 after removing duplicate reads and overlapping bases, after adaptor and quality trimming. Assessment of germline cross-sample contamination was performed using VerifyBamID and samples with more than 3% contamination were excluded. Sex checks were performed to confirm that the sex reported for a participant was concordant with the sex inferred from the genomic data.
WGS alignment and variant calling
COVID-19 cohorts
For the critical and mild COVID-19 cohorts, sequencing data alignment and variant calling were performed with Genomics England pipeline 2.0, which uses the DRAGEN software (v.3.2.22). Alignment was performed to genome reference GRCh38 including decoy contigs and alternative haplotypes (ALT contigs), with ALT-aware mapping and variant calling to improve specificity.
100,000 Genomes Project cohort
All genomes from the 100,000 Genomes Project cohort were analysed with the Illumina North Star Version 4 Whole Genome Sequencing Workflow (NSV4, v.2.6.53.23); which comprises the iSAAC Aligner (v.03.16.02.19) and Starling Small Variant Caller (v.2.4.7). Samples were aligned to the Homo Sapiens NCBI GRCh38 assembly with decoys.
A subset of the genomes from the cancer program of the 100,000 Genomes Project were reprocessed (alignment and variant calling) using the same pipeline used for the COVID-19 cohorts (DRAGEN v.3.2.22) for equity of alignment and variant calling.
Aggregation
Aggregation was conducted separately for the samples analysed with Genomics England pipeline 2.0 (severe cohort, mild cohort, cancer-realigned 100,000 Genomes Project) and those analysed with the Illumina North Star Version 4 pipeline (100,000 Genomes Project).
For the first three, the WGS data were aggregated from single-sample gVCF files to multi-sample VCF files using GVCFGenotyper (GG) v.3.8.1, which accepts gVCF files generated by the DRAGEN pipeline as input. GG outputs multi-allelic variants (several ALT variants per position on the same row), and for downstream analyses the output was decomposed to bi-allelic variants per row using the software vt v.0.57721. We refer to the aggregate as aggCOVID_vX, in which X is the specific freeze.The analysis in this manuscript uses data from freeze v.4.2 and the respective aggregate is referred to as aggCOVID_v4.2.
Aggregation for the 100,000 Genomes Project cohort was performed using Illumina’s gvcfgenotyper v.2019.02.26, merged with bcftools v.1.10.2 and normalized with vt v.0.57721.
Sample quality control
Samples that failed any of the following four BAM-level quality control filters: freemix contamination > 3%, mean autosomal coverage < 25×, per cent mapped reads < 90% or per cent chimeric reads > 5% were excluded from the analysis.
In addition, a set of VCF-level quality control filters were applied after aggregation on all autosomal bi-allelic single-nucleotide variants (SNVs) (akin to gnomAD v.3.1)18. Samples were filtered out on the basis of the residuals of eleven quality control metrics (calculated using bcftools) after regressing out the effects of sequencing platform and the first three ancestry assignment principal components (PCs) (including all linear, quadratic and interaction terms) taken from the sample projections onto the SNP loadings from the individuals of 1000 Genomes Project phase 3 (1KGP3). Samples were removed that were four median absolute deviations (MADs) above or below the median for the following metrics: ratio of heterozygous to homozygous, ratio of insertions to deletions, ratio of transitions to transversions, total deletions, total insertions, total heterozygous SNPs, total homozygous SNPs, total transitions and total transversions. For the number of total singletons (SNPs), samples were removed that were more than 8 MADs above the median. For the ratio of heterozygous to homozygous alternative SNPs, samples were removed that were more than 4 MADs above the median.
After quality control, 79,803 individuals were included in the analysis with the breakdown according to cohort shown in Supplementary Table 2.
Selection of high-quality independent SNPs
We selected high-quality independent variants for inferring kinship coefficients, performing PCA, assigning ancestry and for the conditioning on the genetic relatedness matrix by the logistic mixed model of SAIGE and SAIGE-GENE. To avoid capturing platform and/or analysis pipeline effects for these analyses, we performed very stringent variant quality control as described below.
High-quality common SNPs
We started with autosomal, bi-allelic SNPs which had a frequency of higher than 5% in aggV2 (100,000 Genomes Project participant aggregate) and in the 1KGP3. We then restricted to variants that had missingness < 1%, median genotype quality control > 30, median depth (DP) ≥ 30 and at least 90% of heterozygote genotypes passing an ABratio binomial test with P value > 10−2 for aggV2 participants. We also excluded variants in complex regions from the list available in https://genome.sph.umich.edu/wiki/Regions_of_high_linkage_disequilibrium_(LD) (lifted over for GRCh38), and variants where the REF/ALT combination was CG or AT (C/G, G/C, A/T, T/A). We also removed all SNPs that were out of Hardy–Weinberg equilibrium (HWE) in any of the AFR, EAS, EUR or SAS super-populations of aggV2, with a P value cut-off of PHWE < 10−5. We then LD-pruned using PLINK v.1.9 with r2 = 0.1 and in 500-kb windows. This resulted in a total of 63,523 high-quality sites from aggV2.
We then extracted these high-quality sites from the aggCOVID_v4.2 aggregate and further applied variant quality filters (missingness < 1%, median quality control > 30, median depth ≥ 30 and at least 90% of heterozygote genotypes passing an ABratio binomial test with P value > 10−2), per batch of sequencing platform (that is, HiseqX, NovaSeq6000).
After applying variant filters in aggV2 and aggCOVID_v4.2, we merged the genomic data from the two aggregates for the intersection of the variants, which resulted in a final total of 58,925 sites.
High-quality rare SNPs
We selected high-quality rare (MAF < 0.005) bi-allelic SNPs to be used with SAIGE for aggregate variant testing (AVT) analysis. To create this set, we applied the same variant quality control procedure as with the common variants: We selected variants that had missingness < 1%, median quality control > 30, median depth ≥ 30 and at least 90% of heterozygote genotypes passing an ABratio binomial test with P value > 10−2 per batch of sequencing and genotyping platform (that is, HiSeq + NSV4, HiSeq + Pipeline 2.0, NovaSeq + Pipeline 2.0). We then subsetted those to the following groups of minor allele count (MAC) and MAF categories: MAC 1, 2, 3, 4, 5, 6–10, 11–20, MAC 20–MAF 0.001, MAF 0.001–0.005.
Relatedness, ancestry and principal components
Kinship
We calculated kinship coefficients among all pairs of samples using the software PLINK v.2.0 and its implementation of the KING robust algorithm. We used a kinship cut-off of <0.0442 to select unrelated individuals with argument “–king-cutoff”.
Genetic ancestry prediction
To infer the ancestry of each individual, we performed principal component analysis (PCA) on unrelated 1KGP3 individuals with GCTA v.1.93.1_beta software using high-quality common SNPs43, and inferred the first 20 PCs. We calculated loadings for each SNP, which we used to project aggV2 and aggCOVID_v4.2 individuals onto the 1KGP3 PCs. We then trained a random forest algorithm from the R package randomForest with the first 10 1KGP3 PCs as features and the super-population ancestry of each individual as labels. These were ‘AFR’ for individuals of African ancestry, ‘AMR’ for individuals of American ancestry, ‘EAS’ for individuals of East Asian ancestry, ‘EUR’ for individuals of European ancestry and ‘SAS’ for individuals of South Asian ancestry. We used 500 trees for the training. We then used the trained model to assign a probability of belonging to a certain super-population class for each individual in our cohorts. We assigned individuals to a super-population when class probability ≥ 0.8. Individuals for whom no class had probability ≥ 0.8 were labelled as ‘unassigned’ and were not included in the analyses.
PCA
After labelling each individual with predicted genetic ancestry, we calculated ancestry-specific PCs using GCTA v.1.93.1_beta43. We computed 20 PCs for each of the ancestries that were used in the association analyses (AFR, EAS, EUR and SAS).
Variant quality control
Variant quality control was performed to ensure high quality of variants and to minimize batch effects due to using samples from different sequencing platforms (NovaSeq6000 and HiseqX) and different variant callers (Strelka2 and DRAGEN). We first masked low-quality genotypes setting them to missing, merged aggregate files and then performed additional variant quality control separately for the two major types of association analyses, GWAS and AVT, which concerned common and rare variants, respectively.
Masking
Before any analysis, we masked low-quality genotypes using the bcftools setGT module. Genotypes with DP < 10, genotype quality (GQ) < 20 and heterozygote genotypes failing an ABratio binomial test with P value < 10−3 were set to missing.
We then converted the masked VCF files to PLINK and bgen format using PLINK v.2.0.
Merging of aggregate samples
Merging of aggV2 and aggCOVID_v4.2 samples was done using PLINK files with masked genotypes and the merge function of PLINK v.1.944. for variants that were found in both aggregates.
GWAS analyses
Variant quality control
We restricted all GWAS analyses to common variants applying the following filters using PLINK v.1.9: MAF > 0 in both cases and controls, MAF > 0.5% and MAC > 20, missingness < 2%, differential missingness between cases and controls, mid-P value < 10−5, HWE deviations on unrelated controls, mid-P value < 10−6. Multi-allelic variants were in addition required to have MAF > 0.1% in both aggV2 and aggCOVID_v4.2.
Control–control quality control filter
100,000 Genomes Project aggV2 samples that were aligned and genotype called with the Illumina North Star version 4 pipeline represented the majority of control samples in our GWAS analyses, whereas all of the cases were aligned and called with Genomics England pipeline 2.0 (Supplementary Table 1). Therefore, the alignment and genotyping pipelines partially match the case–control status, which necessitates additional filtering for adjusting for between-pipeline differences in alignment and variant calling. To control for potential batch effects, we used the overlap of 3,954 samples from the Genomics England 100,000 Genomes Project participants that were aligned and called with both pipelines. For each variant, we computed and compared between platforms the inferred allele frequency for the population samples. We then filtered out all variants that had >1% relative difference in allele frequency between platforms. The relative difference was computed on a per-population basis for EUR (n = 3,157), SAS (n = 373), AFR (n = 354) and EAS (n = 81).
Model
We used a two-step logistic mixed model regression approach as implemented in SAIGE v.0.44.5 for single-variant association analyses. In step 1, SAIGE fits the null mixed model and covariates. In step 2, single-variant association tests are performed with the saddlepoint approximation (SPA) correction to calibrate unbalanced case–control ratios. We used the high-quality common variant sites for fitting the null model and sex, age, age2, age-by-sex and 20 PCs as covariates in step 1. The PCs were computed separately by predicted genetic ancestry (that is, EUR-specific, AFR-specific and so on), to capture subtle structure effects.
Analyses
All analyses were done on unrelated individuals with a pairwise kinship coefficient < 0.0442. We conducted GWAS analyses per predicted genetic ancestry, for all populations for which we had more than 100 cases and more than 100 controls (AFR, EAS, EUR and SAS).
Multiple testing correction
As our study is testing variants that were directly sequenced by WGS and not imputed, we calculated the P value significance threshold by estimating the effective number of tests. After selecting the final filtered set of tested variants for each population, we LD-pruned in a window of 250 kb and r2 = 0.8 with PLINK 1.9. We then computed the Bonferroni-corrected P value threshold as 0.05 divided by the number of LD-pruned variants tested in the GWAS. The P value thresholds that were used for declaring statistical significance are provided in Supplementary Table 5.
LD-clumping
We used PLINK v.1.9 to do clumping of variants that were genome-wide significant for each analysis with P1 set to per-population P value from Supplementary Table 5, P2 = 0.01, clump distance 1,500 kb and r2 = 0.1.
Conditional analysis and signal independence
To find the set of independent variants in the per-population analyses, we performed a step-wise conditional analysis with the GWAS summary statistics for each population using GCTA 1.9.3 –cojo-slct function43. The parameters for the function were pval = 2.2 × 10−8, a distance of 10,000 kb and a colinear threshold of 0.9 (ref. 45). For establishing independence of multi-ancestry meta-analysis signals from per-population discovered signals, we performed LD-clumping using the meta-analysis summaries and identified signals with no overlap with the LD-clumped results from the per-population analyses. In addition to the GCTA-cojo analysis, we also performed confirmatory individual-level conditional analysis as implemented in SAIGE. For every lead variant signal (including the multi-ancestry meta-analysis signals), we conditioned on the lead variants of all other signals identified as independent by GCTA-cojo and located on the same chromosome with option –condition of SAIGE (Supplementary Table 6).
Fine-mapping
We performed fine-mapping for genome-wide-significant signals using theR package SusieR v.0.11.4213. For each genome-wide-significant variant locus, we selected the variants 1.5 Mbp on each side and computed the correlation matrix among them with PLINK v.1.9. We then ran the susieR summary-statistics-based function susie_rss and provided the summary z scores from SAIGE (that is, effect size divided by its standard error) and the correlation matrix computed with the same samples that were used for the corresponding GWAS. We required coverage ≥``{=html}0.95 for each identified credible set and minimum and median absolute correlation coefficients (purity) of r = 0.1 and 0.5, respectively.
Functional annotation of credible sets
We annotated all variants included in each credible set identified by SusieR using the online Variant Effect Predictor (VEP) v.104 and selected the worst consequence across GENCODE basic transcripts (Supplementary Information). We also ranked each variant within each credible set according to the predicted consequence and the ranking was based on the table provided by Ensembl: https://www.ensembl.org/info/genome/variation/prediction/predicted_data.html.
Multi-ancestry meta-analysis
We performed a meta-analysis across all ancestries using an inverse-variance weighting method and control for population stratification for each separate analysis in the METAL software46. The meta-analysed variants were filtered for variants with heterogeneity P value P < 2.22 × 10−8 and variants that are not present in at least half of the individuals. We used the meta R package to plot forest plots of the clumped multi-ancestry meta-analysis variants47.
LD-based validation of lead GWAS signals
To quantify the support for genome-wide-significant signals from nearby variants in LD, we assessed the internal consistency of GWAS results of the lead variants and their surroundings. To this end, we compared observed z-scores at lead variants with the expected z-scores based on those observed at neighbouring variants. Specifically, we computed the observed z-score for a variant i as \({s}_{i}=\hat{\beta }/{\hat{\sigma }}_{\hat{\beta }}\) and, following a previous approach48, the imputed z-score at a target variant t as
where \({{\bf{s}}}_{P}\) are the observed z-scores at a set P of predictor variants, \({{\boldsymbol{\Sigma }}}_{x,y}\) is the empirical correlation matrix of dosage coded genotypes computed on the GWAS sample between the variants in x and y, and λ is a regularization parameter set to 10−5. The set P of predictor variants consisted of all variants within 100 kb of the target variant with a genotype correlation with the target variant greater than 0.25. This approach is similar to one proposed recently49.
Stratified analysis
We performed sex-specific analysis (male and female individuals separately) as well as analysis stratified by age (that is, participants of younger than 60 years old and 60 years old or above) for the EUR ancestry group. To compare the effect of variants within groups for the age- and sex-stratified analysis we first adjusted the effect and error of each variant for the standard deviation of the trait in each stratified group and then used the following t-statistic, as in previous studies50,51
where b1 is the adjusted effect for group 1, b2 is the adjusted effect for group 2, se1 and se2 are the adjusted standard errors for groups 1 and 2, respectively, and r is the Spearman rank correlation between groups across all genetic variants.
Replication
To generate a replication set, we conducted a meta-analysis of data from 23andMe, together with a meta-analysis of the COVID-19 HGI data freeze 6 (hospitalized COVID versus population) GWAS (B2 analysis), including all genetic ancestries. Although the HGI programme included an analysis designed to mirror the GenOMICC study (analysis ‘A2’), most of these cases come from GenOMICC and are already included in the discovery cohort. We therefore used the broader hospitalized phenotype (‘B2’) for replication.
To account for signal due to sample overlap we performed a mathematical subtraction from HGI v.6 B2, of the GenOMICC GWAS of European genetic ancestry. Publicly available HGI data were downloaded from https://www.covid19hg.org/results/r6/. The subtraction was performed using the MetaSubtract package (v.1.60) for R (v.4.0.2) after removing variants with the same genomic position and using the lambda.cohorts with genomic inflation calculated on the GenOMICC summary statistics.
We calculated a multi-ancestry meta-analysis for the three ancestries with summary statistics in 23andMe—African, Latino and European—using variants that passed the 23andMe ancestry quality control, with imputation score > 0.6 and with MAF > 0.005, before performing a final meta-analysis of 23andMe and HGI B2 without GenOMICC to create the final replication set. Meta-analysis was performed using METAL46, with the inverse-variance weighting method (STDERR mode) and genomic control ON. We considered that a hit was replicated if the direction of effect in the GenOMICC-subtracted HGI summary statistics was the same as in our GWAS, and the P value was significant after Bonferroni correction for the number of attempted replications (pval < 0.05/25). If the main hit was not present in the HGI–23andMe meta-analysis or if the hit was not replicating, we looked for replication in variants in high LD with the top variant (r2 > 0.9), which helped replicate two regions.
To attempt additional replication of two associations, we performed a multi-ancestry meta-analysis across five continental ancestry groups in the UK Biobank, AncestryDNA, Penn Medicine Biobank and GHS, totalling 9,937 hospitalized cases of COVID-19 and 1,059,390 controls (COVID-19 negative or unknown). Hospitalization status (positive, negative or unknown) was determined on the basis of COVID-19-related ICD10 codes U071, U072, U073 in variable ‘diag_icd10’ (table ‘hesin_diag’) in the UK Biobank study; self-reported hospitalization due to COVID-19 in the AncestryDNA study; and medical records in the GHS and Penn Medicine Biobank studies. Association analyses in each study were performed using the genome-wide Firth logistic regression test implemented in REGENIE. In this implementation, Firth’s approach is applied when the P value from a standard logistic regression score test is less than 0.05. We included in step 1 of REGENIE (that is, prediction of individual trait values based on the genetic data) directly genotyped variants with MAF > 1%, missingness < 10%, HWE test P > 1 × 10−15 and LD-pruning (1,000 variant windows, 100 variant sliding windows and r2 < 0.9). The association model used in step 2 of REGENIE included as covariates age, age2, sex, age-by-sex, and the first 10 ancestry-informative PCs derived from the analysis of a stricter set of LD-pruned (50 variant windows, 5 variant sliding windows and r2 < 0.5) common variants from the array (imputed for the GHS study) data. Within each study, association analyses were performed separately for five different continental ancestries defined on the basis of the array data: African (AFR), Hispanic or Latin American (HLA), East Asian (EAS), European (EUR) and South Asian (SAS). Results were subsequently meta-analysed across studies and ancestries using an inverse-variance-weighted fixed-effects meta-analysis.
HLA imputation and association analysis
HLA types were imputed at two-field (four-digit) resolution for all samples within aggV2 and aggCOVID_v4.2 for the following seven loci: HLA-A, HLA-C, HLA-B, HLA-DRB1, HLA-DQA1, HLA-DQB1 and HLA-DPB1, using the HIBAG package in R15. At the time of writing, HLA types were also imputed for \(\mathop{8}\limits^{ \sim }\) 2% of samples using HLA*LA52. Inferred HLA alleles between HIBAG and HLA*LA were more than 96% identical at four-digit resolution. HLA association analysis was run under an additive model using SAIGE, in an identical manner to the SNV GWAS. The multi-sample VCF of aggregated HLA type calls from HIBAG was used as input in cases in which any allele call with posterior probability (T) < 0.5 were set to missing.
AVT
AVT on aggCOVID_v4.2 was performed using SKAT-O as implemented in SAIGE-GENE v.0.44.517 on all protein-coding genes. Variant and sample quality control for the preparation and masking of the aggregate files have been described elsewhere. We further excluded SNPs with differential missingness between cases and controls (mid-P value < 10−5) or a site-wide missingness above 5%. Only bi-allelic SNPs with MAF < 0.5% were included.
We filtered the variants to include in the AVT by applying two functional annotation filters: a putative loss of function (pLoF) filter, in which only variants that are annotated by LOFTEE18 as high-confidence loss of function were included; and a more lenient (missense) filter, in which variants that have a consequence of missense or worse as annotated by VEP, with a CADD_PHRED score of ≥10, were also included. All variants were annotated using VEP v99. SAIGE-GENE was run with the same covariates used in the single variant analysis: sex, age, age2, age-by-sex and 20 (population-specific) PCs generated from common variants (MAF ≥ 5%).
We ran the tests separately by genetically predicted ancestry, as well as across all four ancestries as a mega-analysis. We considered a gene-wide-significant threshold on the basis of the genes tested per ancestry, correcting for the two masks (pLoF and missense; Supplementary Table 14).
Post-GWAS analysis
TWASs
We performed TWASs in the MetaXcan framework and the GTEx v.8 eQTL and splicing quantitative trait loci (sQTL) MASHR-M models available for download in http://predictdb.org/. We first calculated, using the European summary statistics, individual TWASs for whole blood and lung with the S-PrediXcan function53,54. Then we performed a metaTWAS including data from all tissues to increase statistical power using s-MultiXcan55. We applied the Bonferroni correction to the results to choose significant genes and introns for each analysis.
Colocalization analysis
Significant genes from the TWAS, splicing TWAS, metaTWAS and splicing metaTWAS, as well as genes for which one of the top variants was a significant eQTL or sQTL, were selected for a colocalization analysis using the coloc R package56. We chose the lead SNPs from the European ancestry GWAS summary statistics and a region of ±200 kb around each SNP to do the colocalization with the identified genes in the region. GTEx v.8 whole-blood and lung tissue summary statistics and eqtlGen (which has blood eQTL summary statistics for more than 30,000 individuals) were used for the analysis22,57. We first performed a sensitivity analysis of the posterior probability of colocalization (PPH4) on the prior probability of colocalization (P12), going from P12 = 10−8 to P12 = 10−4, with the default threshold being P12 = 10−5. eQTL signal and GWAS signals were deemed to colocalize if these two criteria were met: (1) at P12 = 5 × 10−5 the probability of colocalization PPH4 > 0.5; and (2) at P12 = 10−5 the probability of independent signal (PPH3) was not the main hypothesis (PPH3 < 0.5). These criteria were chosen to allow eQTLs with weaker P values, owing to lack of power in GTEx v.8, to be colocalized with the signal when the main hypothesis using small priors was that there was not any signal in the eQTL data.
As the chromosome 3-associated interval is larger than 200 kb, we performed additional colocalization including a region up to 500 kb, but no further colocalizations were found.
Mendelian randomization
We performed GSMR23 in a replicated outcome study design. As exposures, we used the pQTLs from the INTERVAL study24. We used the 1000 Genomes Project imputed data of the Health and Retirement Study (HRS) (n = 8,557) as the LD reference data required for GSMR analysis. The HRS data are available from dbGap (accession number: phs000428).
GSMR was undertaken using all exposures for which we were able to identify two or more independent SNPs associated with the exposure (P value(exposure) < 5 × 10−8; LD clumping ±1 Mb, r2 < 0.05; HEIDI-outlier filtering test, for the removal of SNPs with evidence of horizontal pleiotropy, was performed at the default threshold value of 0.01). Using GSMR, we identified those proteins implicated in determining COVID-19 severity in the new GenOMICC results (following genomic-control correction for inflation) at a false discovery rate (FDR) of less than 0.05, and attempted replication in the GWAS of ‘Hospitalized COVID versus population’ (phenotype B2) of the COVID-19 HGI (ref. 58) having excluded the previous GenOMICC results. We achieved this by mathematically removing the contribution of GenOMICC1 from the meta-analysis. We considered as replicated those results that passed a Bonferroni-corrected P value threshold, correcting for the total number of replication tests attempted (that is, the number of observations from the discovery set with FDR < 0.05).
Heritability
For the SNP-based narrow-sense heritabilities of severe COVID-19 and HGI COVID phenotypes, both high-definition likelihood (HDL) and LD score regression (LDSC)59 methods were applied. The HGI summary statistics were based on the GWAS analysis of all available samples, in which the majority were European populations (see https://www.covid19hg.org/results/r6/). The munge_sumstats.py procedure in the LDSC software was used to harmonize the summary statistics, and in LDSC, the reference panel was built using the 1000 Genome European samples with SNPs that have MAF > 0.05. As both HDL and LDSC are based on GWAS summary z-score statistics, the estimated heritabilities are thus on the observed scale.
Enrichment analysis
Enrichment analysis was performed to identify ontologies in which discovery genes were overrepresented. Using the XGR algorithm (http://galahad.well.ox.ac.uk/XGR)60, 19 genes identified through lead variant proximity, credible variant sets, mutation consequence and TWAS analyses were tested for enrichment in disease ontology61, gene ontologies (biological process, molecular function and cellular component)62 and KEGG63 and Reactome64 pathways using default settings. This generated a P value and FDR for overrepresentation of genes within each of the ontologies (Supplementary Table 15).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data are available through https://genomicc.org/data. This includes downloadable summary data tables and instructions for applying to access individual-level data. Individual-level genome sequence data for the COVID-19 severe and mild cohorts can be analysed by qualified researchers in the UK Outbreak Data Analysis Platform at the University of Edinburgh by application at https://genomicc.org/data. Genomic data for the 100,000 Genomes Project participants and a subset of COVID-19 cases are also available through the Genomics England research environment, which can be accessed by application at https://www.genomicsengland.co.uk/join-a-gecip-domain. The full GWAS summary statistics for the 23andMe discovery dataset are available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. More information and access to the data are provided at https://research.23andMe.com/dataset-access/.
Code availability
Code to calculate the imputation of P values based on LD SNPs is available at https://github.com/baillielab/GenOMICC_GWAS.
References
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
Ellinghaus, D. et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534 (2020).
COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 600, 472–477 (2021).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Docherty, A. B. et al. Features of 20,133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ 369, m1985 (2020).
Dorward, D. A. et al. Tissue-specific immunopathology in fatal COVID-19. Am. J. Respir. Crit. Care Med. 203, 192–201 (2021).
Millar, J. E. et al. Distinct clinical symptom patterns in patients hospitalised with COVID-19 in an analysis of 59,011 patients in the ISARIC-4C study. Sci. Rep. 12, 6843 (2022).
The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 384, 693–704 (2021).
Degenhardt, F. et al. New susceptibility loci for severe COVID-19 by detailed GWAS analysis in European populations. Preprint at medRxiv https://doi.org/10.1101/2021.07.21.21260624 (2021).
Povysil, G. et al. Rare loss-of-function variants in type i IFN immunity genes are not associated with severe COVID-19. J. Clin. Invest. 131, e147834 (2021).
Kosmicki, J. A. et al. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am. J. Hum. Genet. 108, 1350–1355 (2021).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B 82, 1273–1300 (2020).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2018).
Zheng, X. et al. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014).
Langton, D. J. et al. The influence of HLA genotype on the severity of COVID-19 infection. HLA 98, 14–22 (2021).
Zhou, W. et al. Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nat. Genet. 52, 634–639 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Asano, T. et al. X-linked recessive TLR7 deficiency in ∼1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6, eabl4348 (2021).
Fallerini, C. et al. Association of toll-like receptor 7 variants with life-threatening COVID-19 disease in males: findings from a nested case-control study. eLife 10, e67569 (2021).
van der Made, C. I. et al. Presence of genetic variants among young men with severe COVID-19. J. Am. Med. Assoc. 324, 663–673 (2020).
The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Dunning, J. W. et al. Open source clinical science for emerging infections. Lancet Infect. Dis. 14, 8–9 (2014).
Dendrou, C. A. et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 8, 363ra149 (2016).
Dong, B. et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J. Virol. 78, 8983–8993 (2004).
Luo, W. et al. Phospholipid scramblase 1 interacts with influenza a virus NP, impairing its nuclear import and thereby suppressing virus replication. PLoS Pathog. 14, e1006851 (2018).
WHO Solidarity Trial Consortium. Repurposed antiviral drugs for Covid-19—interim WHO Solidarity trial results. N. Engl. J. Med. 384, 497–511 (2021).
Kalil, A. C. et al. Efficacy of interferon beta-1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID-19: a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Respir. Med. 12, 1365–1376 (2021).
Yu, Y. et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 209, 2467–2483 (2012).
Reizis, B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity 50, 37–50 (2019).
Zhang, Y., Lu, L., Furlonger, C., Wu, G. E. & Paige, C. J. Hemokinin is a hematopoietic-specific tachykinin that regulates b lymphopoiesis. Nat. Immunol. 1, 392–397 (2000).
Wang, W. et al. Hemokinin-1 activates the MAPK pathway and enhances B cell proliferation and antibody production. J. Immunol. 184, 3590–3597 (2010).
Janelsins, B. M. et al. Proinflammatory tachykinins that signal through the neurokinin 1 receptor promote survival of dendritic cells and potent cellular immunity. Blood 113, 3017–3026 (2009).
Thwaites, R. S. et al. Inflammatory profiles across the spectrum of disease reveal a distinct role for GM-CSF in severe COVID-19. Sci. Immunol. 6, eabg9873 (2021).
Lang, F. M., Lee, K. M.-C., Teijaro, J. R., Becher, B. & Hamilton, J. A. GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat. Rev. Immunol. 20, 507–514 (2020).
Reyes, L. et al. A type I IFN, prothrombotic hyperinflammatory neutrophil signature is distinct for COVID-19 ARDS. Wellcome Open Res. 6, 38 (2021).
Lawler, P. R. et al. Therapeutic anticoagulation with heparin in noncritically ill patients with Covid-19. N. Engl. J. Med. 385, 790–802 (2021).
Amraei, R. et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent. Sci. 7, 1156–1165 (2021).
Thépaut, M. et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLoS Pathog. 17, e1009576 (2021).
Silverman, R. H. & Weiss, S. R. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2-5A. J. Interferon Cytokine Res. 34, 455–463 (2014).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Balduzzi, S., Rücker, G. & Schwarzer, G. How to perform a meta-analysis with R: a practical tutorial. Evid. Based Ment. Health 22, 153–160 (2019).
Pasaniuc, B. et al. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics 30, 2906–2914 (2014).
Chen, W. et al. Improved analyses of GWAS summary statistics by reducing data heterogeneity and errors. Nat. Commun. 12, 7117 (2021).
Bernabeu, E. et al. Sex differences in genetic architecture in the UK biobank. Nat. Genet. 53, 1283–1289 (2021).
Winkler, T. W. et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 11, e1005378 (2015).
Dilthey, A. T. et al. HLA*LA—HLA typing from linearly projected graph alignments. Bioinformatics 35, 4394–4396 (2019).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Barbeira, A. N. et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
The COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Fang, H., Knezevic, B., Burnham, K. L. & Knight, J. C. XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med. 8, 129 (2016).
Schriml, L. M. et al. Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res. 40, D940–D946 (2012).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Chen, M.-H. et al. Phospholipid scramblase 1 contains a nonclassical nuclear localization signal with unique binding site in importin α. J. Biol. Chem. 280, 10599–10606 (2005).
Chen, C.-W., Sowden, M., Zhao, Q., Wiedmer, T. & Sims, P. J. Nuclear phospholipid scramblase 1 prolongs the mitotic expansion of granulocyte precursors during G-CSF-induced granulopoiesis. J. Leukocyte Biol. 90, 221–233 (2011).
Thomas, C. et al. Structural linkage between ligand discrimination and receptor activation by type i interferons. Cell 146, 621–632 (2011).
Acknowledgements
We thank the patients and their loved ones who volunteered to contribute to this study at one of the most difficult times in their lives, and the research staff in every intensive care unit who recruited patients at personal risk under challenging conditions. GenOMICC was funded by the Department of Health and Social Care (DHSC), Illumina, LifeArc, the Medical Research Council (MRC), UKRI, Sepsis Research (the Fiona Elizabeth Agnew Trust), the Intensive Care Society, a Wellcome Trust Senior Research Fellowship (J.K.B., 223164/Z/21/Z) a BBSRC Institute Program Support Grant to the Roslin Institute (BBS/E/D/20002172, BBS/E/D/10002070 and BBS/E/D/30002275) and UKRI grants MC_PC_20004, MC_PC_19025, MC_PC_1905 and MRNO2995X/1. WGS was performed by Illumina at Illumina Laboratory Services and was overseen by Genomics England. We would like to thank all at Genomics England who have contributed to the sequencing, clinical and genomic data analysis. This research is supported in part by the Data and Connectivity National Core Study, led by Health Data Research UK in partnership with the Office for National Statistics and funded by UK Research and Innovation (grant ref. MC_PC_20029). A.D.B. would like to acknowledge funding from the Wellcome PhD training fellowship for clinicians (204979/Z/16/Z) and the Edinburgh Clinical Academic Track (ECAT) programme. We thank the research participants and employees of 23andMe for making this work possible. Genomics England and the 100,000 Genomes Project were funded by the National Institute for Health Research, the Wellcome Trust, the MRC, Cancer Research UK, the DHSC and NHS England. We are grateful for the support from S. Hill and the team in NHS England and the 13 Genomic Medicine Centres that delivered the 100,000 Genomes Project, which provided most of the control genome sequences for this study. We thank the participants in the 100,000 Genomes Project, who made this study possible, and the Genomics England Participant Panel for their strategic advice, involvement and engagement. We acknowledge NHS Digital, Public Health England and the Intensive Care National Audit and Research Centre, who provided life-course longitudinal clinical data on the participants. This work forms part of the portfolio of research of the National Institute for Health Research Barts Biomedical Research Centre. Mark Caulfield is an NIHR Senior Investigator. This study owes a great deal to the National Institute for Healthcare Research Clinical Research Network (NIHR CRN) and the Chief Scientist’s Office (Scotland), who facilitate recruitment into research studies in NHS hospitals, and to the global ISARIC and InFACT consortia. Additional replication was conducted using the UK Biobank Resource (project 26041). The Penn Medicine BioBank is funded by a gift from the Smilow family; the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878; and the Perelman School of Medicine at the University of Pennsylvania. We thank the AncestryDNA customers who voluntarily contributed information in the COVID-19 survey. HRS (dbGaP accession: phs000428.v1.p1): HRS was supported by the National Institute on Aging (NIA U01AG009740). The genotyping was funded separately by the National Institute on Aging (RC2 AG036495, RC4 AG039029). Genotyping was conducted by the NIH Center for Inherited Disease Research (CIDR) at Johns Hopkins University. Genotyping quality control and final preparation of the data were performed by the Genetics Coordinating Center at the University of Washington. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 22 August 2021 (GTEx Analysis Release v.8 (dbGaP Accession phs000424.v8.p2). We thank the research participants and employees of 23andMe for making this work possible. A full list of contributors who have provided data that were collated in the HGI project, including previous iterations, is available at https://www.covid19hg.org/acknowledgements. The views expressed are those of the authors and not necessarily those of the DHSC, NHS, Department for International Development (DID), NIHR, MRC, Wellcome Trust or Public Health England.
Author information
Authors and Affiliations
Consortia
Contributions
A.K., E.P.-C., K. Rawlik, A. Stuckey, C.A.O., S.W., T. Malinauskas, Y.W., X.S., K.S.E., B.W., D.R., L.K., M.Z., N.P., J.A.K., J.E.H., A.B., G.R.A., M.A.R.F., A.J., T. Mirshahi, M.O., D.J.R., M.D.R., A.V., J.Y., A.D.B., S.C.H., L. Moutsianas, A.L. and J.K.B. contributed to data analysis. A.K., E.P.-C., K. Rawlik, A. Stuckey, C.A.O., S.W., C.D.R., J.M., A.R., S.C.H., L. Moutsianas and A.L. contributed to bioinformatics. A.K., E.P.-C., K. Rawlik, C.D.R., J.M., D.M., A.N., M.G.S., S.C.H., L. Moutsianas, M.J.C. and J.K.B. contributed to writing and reviewing the manuscript. E.P.-C., K. Rawlik, K.M., S.K., A.F., L. Murphy, K. Rowan, C.P.P., V.V., J.F.W., S.C.H., A.L., M.J.C. and J.K.B. contributed to design. S.W., F.G., W.O., P.G. and S.D. contributed to project management. F.G., W.O., K.M., S.K., P.G., S.D., D.M., A.N., M.G.S., S.S., J.K., T.A.F., M.S.-H., C.S., C.H., P.H., L.L., D. McAuley, H.M., P.J.O., P.E., T.W., A.T., A.F., L. Murphy, K. Rowan, C.P.P., R.H.S., S.C.H. and A.L. contributed to oversight. F.G., W.O., F.M.-C. and J.K.B. contributed to ethics and governance. K.M., A. Siddiq, A.F. and L. Murphy contributed to sample handling and sequencing. A. Siddiq contributed to data collection. T.Z. contributed to sample handing. T.Z., G.E., C.P., D.B. and C.K. contributed to sequencing. L.T. contributed to the recruitment of controls. G.C., P.A., K. Rowan and A.L. contributed to clinical data management. K. Rowan, C.P.P., S.C.H. and J.K.B. contributed to conception. K. Rowan, C.P.P., V.V. and J.F.W. contributed to reviewing the manuscript. M.J.C. and J.K.B. contributed to scientific leadership.
Corresponding authors
Ethics declarations
Competing interests
J.A.K., J.E.H., A.B., G.R.A. and M.A.R.F. are current employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals. Genomics England is a wholly owned Department of Health and Social Care company created in 2013 to work with the NHS to introduce advanced genomic technologies and analytics into healthcare. All Genomics England affiliated authors are, or were, salaried by Genomics England during this programme. All other authors declare that they have no competing interests relating to this work.
Peer review
Peer review information
Nature thanks Jacques Fellay and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Analysis workflow for GWAS and AVT analyses of this study.
The cohorts displayed in yellow and green in the top box were processed with Genomics England Pipeline 2.0 and Illumina NSV4, respectively (see Methods on WGS Alignment and variant calling for details on differences between pipelines). We used individuals that were processed with either pipeline for the GWAS analyses and individuals processed only with Genomics England Pipeline 2.0 for the AVT analyses. The definition of the cases and controls was the same for GWAS and AVT, cases were the COVID-19 severe individuals for both, and controls included individuals from the 100,000 Genomes Project (100,000 Genomes Project) and also COVID-19 positive individuals that were recruited for this study and experienced only mild symptoms (COVID-mild).
Extended Data Fig. 2 Regional detail showing fine-mapping to identify three adjacent independent signals on chromosome 1.
Top two panels: variants in LD with the lead variants shown. The variants that are included in two independent credible sets are displayed with black outline circles. \({r}^{2}\) values in the legend denote upper limits, 0.2=[0,0.2], 0.4=[0.2,0.4], 0.6=[0.4,0.6], 0.8=[0.6,0.8],1=[0.8,1]. Bottom panel: locations of protein-coding genes, coloured by TWAS \(P\)-value. The red dashed line shows the Bonferroni-corrected \(P\)-value=\(2.2\times {10}^{-8}\) for Europeans.
Extended Data Fig. 3 Regional detail showing fine-mapping to identify two adjacent independent signals on chromosome 19.
Top two panels: variants in LD with the lead variants shown. The variants that are included in two independent credible sets are displayed with black outline circles. \({r}^{2}\) values in the legend denote upper limits, 0.2=[0,0.2], 0.4=[0.2,0.4], 0.6=[0.4,0.6], 0.8=[0.6,0.8],1=[0.8,1]. Bottom panel: locations of protein-coding genes, coloured by TWAS \(P\)-value. The red dashed line shows the Bonferroni-corrected \(P\)-value=\(2.2\times {10}^{-8}\) for Europeans.
Extended Data Fig. 4 Regional detail showing fine-mapping to identify three adjacent independent signals on chromosome 21.
Top three panels: variants in LD with the lead variants shown. The variants that are included in three independent credible sets are displayed with black outline circles. \({r}^{2}\) values in the legend denote upper limits, 0.2=[0,0.2], 0.4=[0.2,0.4], 0.6=[0.4,0.6], 0.8=[0.6,0.8],1=[0.8,1]. Bottom panel: locations of protein-coding genes, coloured by TWAS \(P\)-value. The red dashed line shows the Bonferroni-corrected \(P\)-value=\(2.2\times {10}^{-8}\) for Europeans.
Extended Data Fig. 5 Predicted structural consequences of lead variants at PLSCR1 and IFNA10.
(a) Crystal structure of PLSCR1 nuclear localization signal (orange, Gly257–Ile266, numbering correspond to UniProt entry O15162) in complex with Importin \(\alpha \) (blue), Protein Data Bank (PDB) ID 1Y2A (ref. 65). Side chains of PLSCR1 are shown as connected spheres with carbon atoms coloured in orange, nitrogens in blue and oxygens in red. Hydrogen atoms were not determined at this resolution (2.20) and are not shown. (b) Close-up view showing side chains of PLSCR1 Ser260, His262 and Importin Glu107 as sticks. Distance (in) between selected atoms (PLSCR1 His262 \(N\epsilon 2\) and Importin Glu107 carboxyl O) is indicated. A hydrogen bond between PLSCR1 His262 and Importin Glu107 is indicated with a dashed line. The risk variant is predicted to eliminate this bond, disrupting nuclear import, an essential step for effect on antiviral signalling27 and neutrophil maturation66. (c) Because there is very strong sequence conservation between IFNA10 and the gene encoding IFN\(\omega \), we used existing crystal structure data (Protein Data Bank ID 3SE4 (ref. 67)) for IFN\(\omega \) (cyan) to display a ternary complex with interferon α/β receptor IFNAR1 (blue), IFNAR2 (red). The side chain of Trp164 is shown as spheres and indicated with a black line. (d) The hydrophobic core of IFN\(\omega \) with Trp164 shielded from the solvent in the center. Trp164-surrounding residues of IFN\(\omega \) are numbered and correspond to UniProt entry P05000. Trp164 and surrounding residues are conserved in IFNA10 (UniProt ID P01566) and share the same numbering as in IFN\(\omega \) (P05000). Side chains of four residues are shown as sticks. Carbon and nitrogen atoms coloured in cyan and blue, respectively. The critical COVID-19-associated mutation, Trp164Cys, would replace an evolutionarily conserved, bulky side chain in the hydrophobic core of IFNA10 with a smaller one, which may destabilize IFNA10.
Extended Data Fig. 6 Manhattan plot of HLA and GWAS signal across the extended MHC region for the EUR cohort.
Grey circles mark the GWAS (small variant) associations and diamonds represent the HLA each allele association, coloured by locus. The lead variant from the GWAS and lead allele from HLA are labelled. The left-panel shows the raw association −log10(P values) per variant - prior to conditional analysis. The right-panel shows the −log10(P values) per variant following conditioning on DRB1*04:01. The dashed red line shows the Bonferroni-corrected genome-wide significance threshold for Europeans.
Extended Data Fig. 7 Effect–effect plots for Mendelian randomization analyses to assess causal evidence for circulating proteins in critical COVID-19.
Each plot shows effect size (β) of variants associated with protein concentration (x axis) and critical COVID-19 (y axis). A full list of instruments is found in Supplementary Table 13.
Supplementary information
Supplementary Information
This file contains Supplementary Figures, Supplementary Tables and Supplementary References
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kousathanas, A., Pairo-Castineira, E., Rawlik, K. et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 607, 97–103 (2022). https://doi.org/10.1038/s41586-022-04576-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04576-6
This article is cited by
-
Priority index for critical Covid-19 identifies clinically actionable targets and drugs
Communications Biology (2024)
-
Next-generation sequencing of host genetics risk factors associated with COVID-19 severity and long-COVID in Colombian population
Scientific Reports (2024)
-
Potential use of sodium glucose co-transporter 2 inhibitors during acute illness: a systematic review based on COVID-19
Endocrine (2024)
-
COVID-19 annual update: a narrative review
Human Genomics (2023)
-
Integrative multi-omics approach for identifying molecular signatures and pathways and deriving and validating molecular scores for COVID-19 severity and status
BMC Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
