
Abstract
Since the first cases of COVID-19 were documented in Wuhan, China in 2019, the world has witnessed a devastating global pandemic, with more than 238 million cases, nearly 5 million fatalities and the daily number of people infected increasing rapidly. Here we describe the currently available data on the emergence of the SARS-CoV-2 virus, the causative agent of COVID-19, outline the early viral spread in Wuhan and its transmission patterns in China and across the rest of the world, and highlight how genomic surveillance, together with other data such as those on human mobility, has helped to trace the spread and genetic variation of the virus and has also comprised a key element for the control of the pandemic. We pay particular attention to characterizing and describing the international spread of the major variants of concern of SARS-CoV-2 that were first identified in late 2020 and demonstrate that virus evolution has entered a new phase. More broadly, we highlight our currently limited understanding of coronavirus diversity in nature, the rapid spread of the virus and its variants in such an increasingly connected world, the reduced protection of vaccines, and the urgent need for coordinated global surveillance using genomic techniques. In summary, we provide important information for the prevention and control of both the ongoing COVID-19 pandemic and any new diseases that will inevitably emerge in the human population in future generations.
Similar content being viewed by others

Main
On 31 December 2019, the Wuhan Municipal Health Commission reported an outbreak of pneumonia on its official website. Subsequently, scientists reported the discovery of a previously undescribed coronavirus obtained from samples of the respiratory system of some of these patients. This virus differed from all known coronaviruses including severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS) coronavirus (MERS-CoV)1,2,3,4,5. The World Health Organization (WHO) named the disease coronavirus disease 2019 (COVID-19) and the International Committee on Taxonomy of Viruses named this new infectious agent SARS-CoV-2 (ref. 6); the seventh coronavirus that can infect humans. SARS-CoV-2 rapidly spread through the globally, producing several variants of concern (VOCs) and developing into a major and devastating pandemic. Here we summarize our current understanding of the emergence, global spread and genetic diversity of SARS-CoV-2.
The emergence of SARS-CoV-2
SARS-CoV-2 related coronaviruses
Many of the early cases of COVID-19 in Wuhan, China, were associated with the Huanan Seafood Market2, which—because of the presence of wildlife at the market—was considered an obvious candidate for the location of the initial zoonotic (that is, cross-species transmission) event. However, none of the animals from the market (including rabbits, snakes, stray cats, badgers and bamboo rats) tested positive for SARS-CoV-2 (ref. 7), and viral genome sequences of environmental samples from the market were not considered to occupy basal positions on the viral phylogeny (although the position of the rooting on the tree is uncertain)8. In addition, some of the early cases of COVID-19 in Wuhan were not epidemiologically linked to the market9, and some were linked to other markets10,11. Therefore, although it has not been resolved fully, the current evidence suggests that the Huanan Seafood Market could be the location of an early ‘superspreading’ event.
From the earliest genomic comparisons, it was clear that SARS-CoV-2 had a genomic organization similar to SARS-CoV2. The spike proteins of both viruses have similar three-dimensional structures, suggesting that these viruses might use the same cell surface receptor—human angiotensin-converting enzyme 2(ACE2)2: this was soon confirmed in vitro4,12 and using structural biology12,13. However, SARS-CoV-2 differs from SARS-CoV in two fundamental ways14. First, there are six amino acid positions in the receptor-binding domain (RBD) of the spike protein that mediate the attachment of the SARS-CoV and SARS-CoV-2 spike proteins to the human ACE2 receptor15. However, amino acids at five of the six positions differed between SARS-CoV and SARS-CoV-2 (refs. 2,14). Notably, such differences caused SARS-CoV-2 to have a higher binding avidity to the human ACE2 receptor11, and may have contributed to the higher transmissibility of SARS-CoV-2 compared with SARS-CoV. Second, there is a 12-nucleotide (nt) insertion at the cleavage site of the spike protein of SARS-CoV-2 that has not yet been identified in closely related betacoronaviruses, but that has a complex evolutionary history across the coronaviruses as a whole, indicating that it is evolutionarily volatile16. This insertion encodes four amino acids—PRRA—that can be recognized by the protease furin, which is extensively expressed in different tissues and organs17. This insertion may decrease the overall stability of the SARS-CoV-2 spike, thereby facilitating the adoption of the open conformation that is required for the binding of the spike to human ACE2 (ref. 18); SARS-CoV-2 without this furin-cleavage site shows reduced replication in a human respiratory cell line and was attenuated in laboratory animals19. Notably, amino acid substitutions have been documented at all four positions in the PRRA motif, with a P-to-H substitution (HRRA) identified in more than 487,000 viral genomes as of June 2021.
SARS-CoV-2—like many other members of the genus Betacoronavirus (including SARS-CoV) in the Coronaviridae family—seemingly has its evolutionary roots in those viruses that commonly infect bats2. Not surprisingly, shortly after the identification of SARS-CoV-2, a close relative of SARS-CoV-2 was described; RaTG13 was identified from a bat (Rhinolophus affinis) sample obtained in Yunnan Province, China, in 2013 (ref. 4). Notably, this sample was collected from a mine cave to which four workers were sent to clean bat faeces and who subsequently developed severe pneumonia20. Although RaTG13 exhibits 96.2% sequence identity to SARS-CoV-2 at the scale of the whole genome, it does not possess similar RBD or cleavage-site sequences. Further analyses suggest that RaTG13—rather than SARS-CoV-2—was a recombinant virus, and the two virus lineages probably diverged more than 30 years ago21. Therefore, the SARS-CoV-2 RBD was an ancestral trait shared with bat viruses21.
Subsequently, a number of groups reported the identification of SARS-CoV-2-related coronaviruses in Malayan pangolins (Manis javanica), which were smuggled to Guangxi and Guangdong provinces, China22,23. These pangolin coronavirus genomes exhibited 85.5–92.4% sequence similarity to SARS-CoV-2 (ref. 22). Notably, however, these pangolin-derived coronaviruses formed two sublineages, with the Guangdong sublineage clustering with RaTG13 and SARS-CoV-2 and sharing 97.4% amino-acid similarity to SARS-CoV-2 in the RBD, with identical amino acids at the five critical residues of the RBD. Furthermore, the Guangdong pangolins appeared to have a similar disease manifestation to people with COVID-19 (ref. 24). Therefore, although the role—if any—of pangolins in the origin of SARS-CoV-2 and the ecology of coronaviruses in general is unknown, it is clear that coronaviruses exist in wildlife and that these viruses possess SARS-CoV-2-like RBDs and have a high binding avidity to hACE2.
Furthermore, a previously undescribed bat coronavirus—RmYN02—was reported, which had been collected during routine surveillance of Rhinolophus malayanus bats in Yunnan Province on 25 June 2019 (ref. 25). RmYN02 shared 97.2% sequence identity with SARS-CoV-2 in open-reading frame (ORF) 1ab. ORF1ab is the largest in coronaviruses with a length of approximately 21,300 nt. In June 2021, we reported four SARS-CoV-2-related coronaviruses genomes from Yunnan Province26. Of these, RpYN06, found in Rhinolophus pusillus, exhibited 94.5% sequence identity to SARS-CoV-2. However, the genome—excluding the spike gene, which has a history of recombination—had a similarity to SARS-CoV-2 of 97.2%, making it the closest related genomic backbone to SARS-CoV-2 identified to date. The other three SARS-CoV-2-related coronaviruses were more distantly related to SARS-CoV-2. However, they carried a genetically distinct spike genes encoding proteins that could bind to the human ACE2 receptor in vitro, albeit weakly.
SARS-CoV-2-like coronaviruses have also been identified in bat populations from other parts of Asia, including Japan27, Cambodia28 and Thailand29. Notably, although two betacoronaviruses (STT182 and STT200) from Rhinolophus shameli bats sampled in 2010 from Cambodia shared 92.6% nucleotide identity with SARS-CoV-2 across the genome as a whole, they share five of the six critical RBD sites observed in SARS-CoV-2 and the Guangdong pangolin coronavirus28. In September 2021, a preprint described a number of SARS-CoV-2-related coronaviruses identified in Laos, including BANAL-52 from R. malayanus, BANAL-103 from R. pusillus and BANAL-236 from Rhinolophus marshalli, which only possessed one or two amino acid mismatches at the seventeen residues that interact with human ACE229. In particular, the RBDs of these viruses could bind as efficiently to the human ACE2 protein as could the SARS-CoV-2 Wuhan strain from the early stage of the pandemic.
Emergence pathways of SARS-CoV-2
There are several hypotheses regarding the origin and emergence of SARS-CoV-2 that have been thoroughly clarified in the WHO–China joint report7. These contradictory hypotheses have raised standing debates, with the central point being two competing hypotheses: zoonotic emergence (including direct zoonotic introduction or introduction through an intermediate host) and a laboratory escape. The discovery of more and more SARS-CoV-2-related coronaviruses from wild animals provides evidence for a zoonotic origin of SARS-CoV-2 (refs. 4,22,23,25,26,27,28,29,30). Notably, all of the SARS-CoV-2-related coronaviruses mentioned above are evidently not the direct ancestor of SARS-CoV-2. Any such direct ancestral virus—which has yet to be identified—would be expected to exhibit more than 99% similarity to SARS-CoV-2 across the genome as a whole. However, the discovery of these viruses again highlights that more-closely related viruses in bats and other wildlife species will be identified with enhanced sampling in a broader geographical region, including most parts of Southeast Asia, which has a high diversity of Rhinolophus species26. As it has seldomly been found that a bat coronavirus is able to efficiently transmit among humans without adaptation and repeated human–animal contacts10, introduction through an intermediate host, such as raccoon dogs, is more likely than a direct zoonotic introduction.
Whether SARS-CoV-2 was introduced through a laboratory accident or whether it has been genetically manipulated is highly debatable. After a thorough analysis of the genetic characterizations of SARS-CoV-2 from both the early and later stages of the pandemic, as well as its close relatives from wild animals, many researchers in the global scientific community have reached the consensus that SARS-CoV-2 is unlikely to have escaped a laboratory and there is no scientific evidence that SARS-CoV-2 has been genetically manipulated10. However, the exact spillover event and emergence process of SARS-CoV-2 is still unclear, and more information from the earliest stage of the epidemic is clearly important to understand how SARS-CoV-2 came into contact with people.
Global genetic diversity of SARS-CoV-2
Genomic surveillance of SARS-CoV-2
Mutations are a natural part of the replication cycle of any RNA virus, leading to the diversification of viral lineages when coupled with inter-host transmission. This is also true for SARS-CoV-2, even though coronaviruses contain certain proofreading mechanisms that enhance genome fidelity31. Genomic surveillance has generated an unprecedented amount of sequencing data for a single virus (Box 1), and has proven an essential tool32,33 for tracing the spread of SARS-CoV-2 at various scales, from individual transmission events to the intercontinental spread of the virus. In addition, it has had a central role in monitoring the evolution of SARS-CoV-2 and identifying new variants with enhanced transmissibility and/or pathogenicity, decreased susceptibility to therapeutic agents and that are capable of evading natural or vaccine-induced immunity (Fig. 1). Genomic surveillance has demonstrated the effectiveness of tracking local transmission events, recognizing importation sources and superspreading events in Australia34,35, for informing public-health decision-making in the Netherlands36, and for adopting social-distancing measures to reduce viral spread in Israel37. In January 2021, du Plessis and colleagues described the analysis of 50,887 SARS-CoV-2 genomes38, quantifying the viral genetic structure of the UK epidemic at a fine scale, including the size, spatiotemporal origins and persistence of lineages as well as the effect of intervention measures.

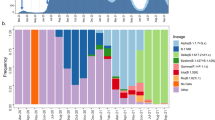
The phylogenetic analysis was performed using full-length genome sequences of SARS-CoV-2 collected from GISAID as of 12 May 2021. A maximum likelihood tree of 1,715 representative high-quality SARS-CoV-2 sequences carrying specific accumulative mutations was estimated using RAxML159, with 1,000 bootstrap replicates and the GTR nucleotide substitution model. The major VOCs (Alpha to Delta) are shown in orange, and the major variants of interests (Epsilon to Lambda) are shown in purple. Both the thickness of each branch in the phylogenetic tree and the shading from light to dark in the heat map indicate the number of sequences carrying specific sets of mutations. Specific nucleotide substitutions are highlighted on the major branches of the tree. The branches with the D614G substitution are coloured blue.
Below, we use Guangdong Province, China and the USA as examples to illustrate how genomic surveillance has facilitated our understanding of this pandemic.
Guangdong, China
Guangdong is a populous province in Southeast China, with a resident population of more than 100 million people. After the SARS-CoV outbreak, believed to have originated in Guangdong39, long-term reforms in public-health agencies have greatly improved the infrastructures and enhanced the capacity of disease control and prevention. The first case of COVID-19 in Guangdong had an onset of symptom on 1 January and was reported on 19 January 2020 (refs. 11,40). Like many other Chinese provinces, Guangdong experienced three phases—domestic importation, local community transmission and international importation—with an epidemic peak in early February 2020 (ref. 40). Large-scale surveillance (around 1.6 million tests by 19 March 2020 identifying 1,388 cases of COVID-19) and intervention measures were implemented from the beginning of the outbreak, and after 22 February 2020 no more than one case a day was reported40. The genomic epidemiology of SARS-CoV-2 in Guangdong showed that most of the infections before March were imported from Hubei Province, and, in particular, Wuhan. Although some early cases were caused by community transmission, local transmission chains were limited both in size and duration40. These results highlight the efficacy of intensive testing and contact tracing even in such a densely populated urban region. Intensive surveillance also identified two SARS-CoV-2 variants with deletions in the spike gene41. In addition, the Guangdong Centers for Disease Control and Prevention (CDC) successfully identified the imported Alpha and Beta variants on 2 January 2021 (ref. 42) and 6 January 2021 (ref. 43), respectively.
The USA
The first case of COVID-19 in the USA (sequence WA1) was reported on 20 January 2020—a traveller from Wuhan44. By 15 February 2020, the number of laboratory-confirmed and clinically diagnosed cases of COVID-19 had reached 15 (ref. 45). By combining multiple sources of information, Worobey and colleagues showed that transmission of the WA1 (belonging to lineage A) lineage was successfully contained, and the subsequent larger outbreaks in Washington state might have been caused by multiple independent introductions of the virus from China in late January or early February 2020 (ref. 46). However, evidence from various studies revealed that the early viruses that were present between 29 February and 18 March 2020 in New York City were imported from Europe and other parts of the USA by multiple, independent introductions47. In addition, cryptic transmission and a prolonged period of unrecognized community spread has been documented in northern California48, Washington state49 and New York City50 from late January to March 2020. For example, SARS-CoV-2 sequences sampled from Connecticut during 6–14 March 2020 group with those from Washington state, highlighting long-distance domestic transmission51. Genomic surveillance in Dane and Milwaukee counties in Wisconsin between March and April 2020 provided evidence for reduced viral spread after a state-wide ‘safer at home’ order52. Together, these genomic surveillance studies clearly illustrate the early transmission of SARS-CoV-2 and highlight the efficacy of intensive testing, contact tracing and decreasing public gatherings in containing the spread of SARS-CoV-2.
Mutational diversity of SARS-CoV-2
By January 2021, approximately 25,000 out of the 29,800 sites (the length of the complete SARS-CoV-2 genome) have been shown to carry mutational differences (https://bigd.big.ac.cn/ncov/), and it has been estimated that approximately two mutations are fixed in the SARS-CoV-2 genome per month46,53,54. Although most of these mutations represent standard replication errors, host-dependent RNA editing may also shape the short- and long-term evolution of SARS-CoV-2. Indeed, the SARS-CoV-2 genome is characterized by frequent biased C-to-U hypermutation that is probably due to a human APOBEC-like editing process55,56.
Similar to other coronaviruses, the spike protein of SARS-CoV-2 contains important antigen epitopes57,58. As such, mutations in the spike protein will probably affect the receptor-binding efficiency, potentially lead to immune escape and may even weaken vaccine efficacy. The first notable mutation was A23403G, which caused the D614G amino acid substitution in the spike protein. This mutation might have arisen separately as early as late January 2020 in China and later in Europe, representing an interesting evolution of a mutation of convergence, and the frequency of this mutation greatly increased during the outbreak in Europe59,60. There is now compelling evidence that D614G has increased virus infectivity and transmissibility59,60,61,62,63,64, and molecular epidemiological studies suggest that this mutation increased the basic reproduction number (R0) from 3.1 (614D) to 4.0 (614G)60. In addition, a so-called ‘cluster V’ (also called B.1.1.298) SARS-CoV-2 variant was identified in Danish mink that also carried mutations in the spike protein, including Y453F, I692V, M1229I and the deletion of two amino acids (Δ69–Δ70)16,65 (Fig. 2).

Three-dimensional structures were modelled with the Swiss-Model program using the spike protein of SARS-CoV-2 (PDB: 7CWU.1.G) as a template. Left, red spheres represent the mutations found in the Alpha160, Beta69, Gamma146 and Delta146 VOCs, as well as the mink cluster V variants65. The amino acid positions of all of the strains are numbered according to the template. Right, the surfaces of the six amino acid residues (L455, F486, Q493, S494, N501 and Y505) at the RBD are coloured cyan. The molecular surfaces of the mutations in the Alpha (purple), Beta (blue), Gamma (yellow), Delta (green), and mink cluster V (pink) variants are highlighted. *Not all Alpha variants have the E484K and S494P mutations. #Not all Delta variants possess the G142D mutation. It should be noted that we only use this figure to highlight the locations of the mutations in the variants based on the three-dimensional structure of one ancestral Wuhan strain (NC_045512), and this figure does not necessarily represent the true three-dimensional structure of the variants.
Not surprisingly, as the number of cases of COVID-19 continued to rise, mutational variants with a likely greater effect on fitness have also emerged, including some that might result in immune escape. Indeed, there are putative escape mutations to the ten human monoclonal antibodies that target the SARS-CoV-2 RBD66. Of particular note are the major SARS-CoV-2 VOCs that arose in late 2020: Alpha (also known as B.1.1.7 and VOC-202012/01), Beta (also known as B.1.351 and 501Y.V2), Gamma (also known as P.1) and Delta (also known as B.1.617.2); these VOCs were first identified in the UK67,68, South Africa69, Brazil70,71 and India72, respectively (Box 2 and Figs. 1, 2).
The emergence of these variant lineages has raised concerns that the virus has entered a new phase in its evolution73,74,75, characterized by ongoing immune escape in the face of increasing levels of infected hosts that probably affects vaccine efficacy, as well as the possibility of selection for increased transmission due to the imposition of nonpharmaceutical interventions (NPIs)74. The Alpha variant has been associated with increased rates of virus population growth67,68 and has been reported to be able to escape neutralization by most monoclonal antibodies targeting the N-terminal domain (NTD) of the spike protein76. However, there is no widespread escape of the Alpha variant from monoclonal antibodies or antibody responses generated by natural infection or vaccination76,77,78, such that its spread may instead reflect increased transmissibility. In particular, some of the Alpha variants acquired additional mutations in the spike protein, especially E484K, and exhibited a substantial loss of sensitivity to the neutralizing activity of vaccine-elicited antibodies and resistance to neutralization by monoclonal antibodies in COVID-19-convalescent plasma79. More worryingly, the Beta variant can escape neutralization by most RBD-targeting monoclonal antibodies and substantially escape from neutralizing antibodies from COVID-19-convalescent plasma76,80,81. Similarly, the Gamma variant shows marked decreases in neutralization with post-vaccination sera82; although, surprisingly, it is considerably less resistant to naturally acquired or vaccine-induced antibody responses than the Beta lineage83. Furthermore, neutralization of the Delta lineage is reduced when compared with ancestral circulating strains77,78, and convalescent sera from patients infected with the Beta and Gamma variants show a markedly higher reduction in neutralization of the Delta lineage77.
In addition to nucleotide substitutions, the SARS-CoV-2 genome has experienced many deletion events. For example, some viruses from Singapore and Taiwan, China carried a 382-nt deletion truncating ORF7b and covering almost the entire ORF8 sequence84,85,86. This variant showed considerably higher replicative fitness in vitro than the wild-type virus84, but seemed to be associated with a milder infection clinically85 and has not been reported in 2021. Su and colleagues described other ORF7b/8 deletions of various lengths, including viruses from Australia (138 nt), Bangladesh (345 nt) and Spain (62 nt)84. Long deletion events were also found in clinical samples from Beijing, with a 120-nt deletion in ORF7a and a 154-nt deletion in ORF8 (ref. 87).
Global spread of SARS-CoV-2
Initial spread of SARS-CoV-2 in China
Generally, China experienced three distinct phases of SARS-CoV-2 transmission: (1) the initial rapid spread in Wuhan; (2) seeding from Wuhan to cause community transmission in other regions of China; and (3) sporadic outbreaks caused by international importations after China controlled the first wave40,87.
Early spread of SARS-CoV-2 in Wuhan
The initial SARS-CoV-2 outbreak in Wuhan can itself be divided into three phases88: (1) rapid transmission before the implementation of the large-scale population ‘lockdown’ of the city on 23 January 2020 (ref. 9), with an estimated effective reproduction number (Re) of 3.5 (95% credible interval, 3.4–3.7) during this period89; (2) reduction of the rate of virus transmission during the period 23 January–1 February 2020 (through lockdown and home quarantine), producing an average Re of 1.2 (95% credible interval, 1.1-1.3)89; and (3) the interruption of transmission through intensified stringent interventions during 2–16 February 2020 (centralized isolation and treatment of cases of COVID-19) and 17 February–8 March 2020 (community screening). Population-based serological surveys conducted during March–May 2020 revealed that the overall seropositivity rate in Wuhan was 3.2–4.4% (refs. 90,91,92,93), indicating that many cases went undetected due to asymptomatic and mild infections and the limited laboratory-diagnosis capacity during the early stages of the outbreak89,94,95. However, city-wide nucleic acid screening of SARS-CoV-2 between 14 May and 1 June 2020 among nearly 10 million residents of Wuhan only found around 300 individuals who had asymptomatic infections after the lockdown was lifted on 8 April 2020 (ref. 96) and no symptomatic local cases related to the initial wave have been reported in the city after 10 May 2020.
Spread from Wuhan to other provinces
The coincidence of the emergence of SARS-CoV-2 and the large-scale seasonal migration (Chunyun, starting from 10 January 2020) for the Chinese Lunar New Year holiday probably exacerbated the seeding of the virus across China97,98. Movement restrictions from Wuhan, the key transportation hub in central China, commenced on 23 January 2020, and reduced the peak population numbers leaving the city 2 days before the Lunar New Year. Unfortunately, however, the disease had spread to every province in mainland China by this time99,100. In general, after the rapid implementation of stringent and integrated NPIs, the Re in provinces outside Hubei decreased below the epidemic threshold (1.0) from 8 February 2020 (ref. 101). Compared with Wuhan, the seropositivity rate in cities outside Wuhan was much lower. According to a national COVID-19 sero-epidemiological survey in China during March–May 2020 (ref. 92), only 0.44% of the sampled population in other cities of Hubei were positive, and only 2 out of more than 12,000 people outside Hubei tested positive, suggesting that SARS-CoV-2 transmission was well contained across the country during the first wave99,102,103.
Frequent international importation events
More than 6,000 incoming travellers from abroad who were infected with SARS-CoV-2 had been reported in mainland China by 15 June 2021, although reverse-transcriptase–polymerase-chain-reaction (RT–PCR) testing at the border control and a 14-day centralized quarantine implemented in China since March 2020 greatly reduced any transmission risk. For example, in Guangzhou, Guangdong Province in southern China, 73.5% of the imported positive cases were detected at the immigration checkpoint and 19.0% during centralized quarantine in hotels104. Although SARS-CoV-2 is predominantly associated with respiratory transmission, since June 2020, multiple Chinese provinces have detected SARS-CoV-2 RNA or live virus on packages of frozen products105. Indeed, cold-chain food or package contamination was proposed to have triggered the resurgence in Beijing in June 2020 (ref. 106) as well as other sporadic outbreaks in China105, although this warrants further investigation. It is notable that the number of confirmed cases was low in the Xinfadi outbreak, Beijing, in June 2020. Similarly, all of the COVID-19 outbreaks in China triggered by international inbound travellers were small-scale, with a few sustained cases. This was mainly due to the citywide, grid-based mass-screening protocol using RT–PCR testing107.
Intercontinental spread of SARS-CoV-2
From China to other regions
The global spread of SARS-CoV-2 shows how rapidly geographically disparate countries can be reached by an emerging pathogen108,109 (Fig. 3a). Two distinct transmission phases of international exportations of SARS-CoV-2 were identified at the early stage of the pandemic110. In the first phase, many international airline passengers left Wuhan for hundreds of destinations across the world during the two weeks before the Wuhan lockdown92. Cities across Asia, Europe and North America were the main destinations and reported several imported cases during the early stage of the COVID-19 outbreak109,111, and the WHO declared a Public Health Emergency of International Concern on 30 January 2020. Containment of the outbreak in China and, in particular, the implementation of travel restrictions since late January 2020 considerably reduced the further spread of SARS-CoV-2 outside China99,100,102,112,113.

a, The date of the first reported case of COVID-19 in each country, territory or area, though the origin of SARS-CoV-2 has not been determined for almost two years. The areas without data are shown in grey. b, Reports of VOCs (now denoted VOC Alpha to Delta) based on records published in the COVID-19 weekly epidemiological update by the WHO (https://covid19.who.int/), as of 10 August 2021. c, The seven-day rolling average of the number of confirmed cases of COVID-19 reported by continent. The orange vertical dashed line indicates the date of COVID-19 declared as a pandemic by the WHO. d, e, The weekly proportion of case number in the top-50 ranked countries with the highest number of cases of COVID-19 (d) and the available mobility data (e), as of 8 August 2021. The weekly proportion was calculated as the case count in a specific week and country, divided by the total number of cases reported in each country. e, The changes in human mobility (by 8 August 2021) in the 50 countries presented in d, compared to the normal mobility from 3 January to 6 February 2020. Each row in d and e represents a country, grouped by continent and then sorted by the latitudes of capital cities from north to south (the country list is available in Supplementary Table 1). The grey dotted vertical lines in d and e from left to right indicate the first week of April, July and October in 2020, and January, April and July in 2021, respectively. The dataset of case numbers was obtained from the data repository collated by the Johns Hopkins University (github.com/CSSEGISandData/COVID-19). The anonymized and aggregated data of population mobility in transit stations were obtained from the Google COVID-19 Community Mobility Reports (www.google.com/covid19/mobility/). The administrative boundary maps were obtained from Natural Earth (www.naturalearthdata.com).
From Europe to other regions
However, international travel outside China from mid-February to late-March 2020 facilitated the second phase of international SARS-CoV-2 spread and onward transmissions110,114, with the epicentre quickly shifting to the Middle East115 and Europe (Fig. 3c). Although France was the first country to identify cases of COVID-19 in Europe, Italy soon became the first major hotspot in the continent111,112,116,117, whereas Spain, Belgium and the UK reported the highest numbers of deaths in Europe during the first wave118. The virus exported from Europe acted as a major source of global spread47, and the WHO eventually declared a pandemic on 11 March 2020. Countries quickly placed restrictions on flights from Europe during March–April 2020, although these measures could not fully prevent introduced transmission75,114.
By late March 2020, cases surged in the USA, with North America becoming the global epicentre119,120. By the end of 2020, the total number of confirmed cases recorded in the USA had passed 20 million, including more than 350,000 reported deaths. Although the first case of COVID-19 in the USA was reported in a traveller returning from China on 20 January 2020 (ref. 44), phylogenetic evidence suggests that importations from Europe mainly contributed to the wide spread of the virus across the country110,119. Latin America and south Asia have also been badly affected. SARS-CoV-2 was confirmed in Brazil on 25 February 2020 and a month later it was found in every state, with confirmed cases exceeding 1 million on 19 June 2020 (refs. 121,122). Although the first case of COVID-19 was confirmed in India on 30 January 2020 and the situation was seemingly under control until the end of March 2020 (ref. 123), India has reported the second highest number of cases of COVID-19 since September 2020 (ref. 124). Most African countries experienced community transmission by 31 May 2020, with most imported cases returning from Europe and the USA125, and it is believed that the disease is generally underreported across Africa due to the limited testing and healthcare capacities126,127,128,129.
Spread of secondary waves across countries
NPIs—such as travel restrictions, case isolation and contact tracing, physical distancing, face covering, hand washing and even the closures of businesses and schools—have been widely implemented to reduce the transmission of SARS-CoV-2 (refs. 112,130,131). Full or partial lockdowns during specific periods have also been imposed in many countries118. Although the effectiveness of different interventions and their combinations have varied, these measures have had an important role in the response to the first wave of the pandemic132,133.
Unfortunately, after the relaxation of these interventions, an increase in population movements and the spread of new variants with a higher transmissibility, a new wave of infections has swept through many nations since October 2020 (refs. 134,135,136) Fig. 3d, e and Supplementary Table 1). The first US wave in 2020 mainly affected the northeast of the USA137, whereas the second wave in summer 2020 mainly hit the south and west, and almost every state has seen a spike in cases during the third wave since October 2020 (ref. 138). Brazil has experienced a major second wave since November 2020 and even had a death toll second only to the USA in early 2021 (ref. 139). New SARS-CoV-2 variants also spread throughout Europe after travel resumed in summer 2020140,141, with the highest daily number of cases recorded in many countries between October 2020 and March 2021. After NPIs were implemented together with a second or third lockdown, and combined with ongoing and large-scale vaccination efforts, many countries passed the second wave by the end of May 2021. This has reduced the pressure on healthcare systems and given countries time to vaccinate people at the greatest risk of severe disease142.
However, the emergence and rapid spread of various SARS-CoV-2 VOCs and variants of interest (VOIs) that are more contagious and/or potentially evade immunity has triggered new waves in many countries (Fig. 3b and Extended Data Fig. 1). For example, India has experienced a major second wave from March to June 2021, mostly due to the Delta variant. As of 10 August 2021, a total of 142 countries, territories and areas across the world have reported the Delta variant72 (Extended Data Fig. 1d), including countries with mass vaccination of their populations, such as the UK and Israel143. In particular, community transmission of this variant has been reported in many countries72. In mid-June 2021, the WHO declared that the Delta variant has displaced most of the other VOCs and has become the dominant lineage across the world143,144.
Challenges and outlook
Even though it is of vital importance to the prevention of future emerging infectious diseases that will inevitably affect human populations, our current understanding of the initial SARS-CoV-2 spillover event is limited. Although the closest relatives to SARS-CoV-2 are found in horseshoe bats, it remains unclear whether the virus directly moved from bats to humans or was passed through an intermediate animal host—as was the case for previous coronavirus epidemics—although the latter seems more reasonable7,10.
The genomic surveillance of SARS-CoV-2 is by far the largest pathogen genomic sequencing project undertaken, with more than 2.8 million complete genomes generated as of August 2021. This endeavour has played an essential part in the prevention and control of COVID-19 and shed light on the transmission patterns of SARS-CoV-2 at different scales, such as the time and source of the introduction events, the spatiotemporal characterizations of local spread, the role of superspreading events, and the viral factors associated with the fitness, transmissibility, infectivity and disease severity. Of particular note is the identification of the major SARS-CoV-2 VOCs, as well as several variants of interests (denoted Epsilon to Lambda)145,146 that emerged in different countries and have caused an increased proportion of cases both locally and globally.
The emergence of these SARS-CoV-2 variants has shaped the complex global transmission dynamics of COVID-19. More importantly, there is mounting evidence147 that these SARS-CoV-2 variants are able to cause decreases in neutralizing titres from patients who recovered from COVID-19 and vaccine recipients, and escape neutralization by the monoclonal antibodies that target the NTD and RBD of the spike protein to various degrees. However, genomic surveillance would be more informative if coupled with a system for the risk assessment and phenotyping of these mutations. For example, the infectivity and antigenicity of 106 mutations in the SARS-CoV-2 spike was assessed using pseudotyped viruses148. Deep mutational scanning has also been used to assess all single amino acid variants of the SARS-CoV-2 spike protein66,149. In addition, more and more data on antigenic variations of the SARS-CoV-2 variants, with different sets of single amino acid mutations, to monoclonal antibodies and vaccines are available. A risk assessment system that integrates pathogen surveillance, immune escape data and near real-time human mobility metrics is desirable, although it may be confounded by the different classes of neutralizing and NTD antibodies, vaccine strategies and even host heterogeneity.
That the major SARS-CoV-2 VOCs have reduced the efficacy of monoclonal antibodies and vaccines has posed serious challenges to the control of the COVID-19 pandemic. First, although vaccines can protect people infected with SARS-CoV-2 variants against severe disease, vaccine manufacturers are exploring redesigns of their products to obtain more effective protection—to eventually prevent virus transmission. Second, the suboptimal protection provided by vaccines150 and the deployment of antibody-based treatments of limited or undemonstrated efficacy151 has raised concerns that this would accelerate the emergence of new variants, although there is a strong argument for mass vaccination even if vaccines can only provide partial immunity152,153. Third, this has also raised the possibility that SARS-CoV-2 will become a recurrent seasonal infection154,155. Fourth, because vaccines cannot completely prevent transmission of the major variants, some NPIs such as face covering might have to be implemented to reduce transmission of the virus, as unlimited, large-scale spread of the variants would probably generate more new variants.
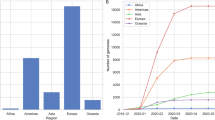
The genomic surveillance of SARS-CoV-2 is also facing several major challenges. First, despite this enormous endeavour, in reality only a very small proportion (around 2%) of cases have been sequenced. In addition, the majority of sequences come from a small number of countries and, remarkably, as of August 2021, around 50% of genomes have been generated in the UK and the USA, which have led the worldwide effort in this respect. By contrast, other countries with major outbreaks, such as India and Brazil, have sequenced a much smaller numbers of cases, which may cause delays in identifying variants with previously undescribed phenotypic characteristics. Therefore, it is likely that there are additional new variants that have not yet been detected given the limited genomic surveillance in a number of regions. Indeed, because the major VOCs are genetically divergent, it is possible that they have been circulating cryptically in unsampled locations, or have also emerged in individuals with a chronically infection who shed the virus for extended periods156,157. Second, the complex transmission dynamics caused by different SARS-CoV-2 variants and their continuous evolution clearly necessitate increased genomic surveillance in such a world with global connectivity and travel networks reshaped by the pandemic. Third, it is possible that recombination among viruses will also change the genetic structure of SARS-CoV-2, perhaps generating viruses with an altered phenotype. Indeed, there have already been suggestions of recombination between the Alpha and Epsilon variants in California in early 2021 (ref. 158). Similarly, the potential recombination of SARS-CoV-2 and other mild human coronaviruses should not be neglected.
In summary, SARS-CoV-2 has led to an increased understanding of coronavirus evolution and the virus has entered a new evolutionary phase characterized by the frequent emergence and spread of variants that affect immune escape and reduce the efficacy of vaccines. Of particular concern is that the limited genomic surveillance in many low-income countries may cause delays in identifying variants with previously undescribed phenotypic characteristics. To contain the current and future pandemics, we urgently call for closer international cooperation, increased vaccine supply and sharing, rapid information exchange, and the establishment of both the infrastructure and trained personnel required for the effective genomic surveillance of SARS-CoV-2 and other emerging viruses.
References
Zhu, N. et al.A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382, 727–733 (2020). One of the first papers to describe the identification of SARS-CoV-2 in Wuhan in late December 2019, providing the sequences of three full-length viral genomes and the successful isolation of the novel coronavirus.
Lu, R. et al.Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020). This study describes the genomic structure and phylogenetic position of eight complte and two partial SARS-CoV-2 genome sequences obtained from samples of nine patients from different hospitals in Wuhan in late December, 2019.
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020). In addition to providing the early identification of SARS-CoV-2, this paper includes a description of the closest relative of SARS-CoV-2 identified to date—the bat-derived coronavirus RaTG13.
Tan, W. et al. A novel coronavirus genome identified in a cluster of pneumonia cases - Wuhan, China 2019–2020. China CDC Weekly 2, 61–62 (2020). Genomic sequencing of the novel coronavirus, initially named after nCoV-19.
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
WHO. WHO-convened Global Study of Origins of SARS-CoV-2: China Part. Joint WHO–China Study (WHO, 2021).
Hill, V. & Rambaut, A. Phylodynamic analysis of SARS-CoV-2. Virological https://virological.org/t/phylodynamic-analysis-of-sars-cov-2-update-2020-03-06/420 (2020).
Li, Q. et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020). An early estimate of the epidemiological characteristics at the start of the COVID-19 outbreak, providing important evidence of human-to-human transmission from the middle of December 2019 in Wuhan, China.
Holmes, E. C. et al. The origins of SARS-CoV-2: a critical review. Cell 184, 4848–4856 (2021).
Chan, J. F. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523 (2020).
Wang, Q. H. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904 (2020). Identification and structural basis of the binding of the ACE2 receptor to SARS-CoV-2.
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020). This paper describes the fundamental genomic features of SARS-CoV-2—in particular, the RBD and the furin cleavage site—and outlines the case for its zoonotic origin.
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127-20 (2020).
Garry, R. F. & Gallaher, W. R. Naturally occurring indels in multiple coronavirus spikes. Virological https://virological.org/t/naturally-occurring-indels-in-multiple-coronavirus-spikes/560 (2020).
Li, X. et al. A furin cleavage site was discovered in the S protein of the 2019 novel coronavirus [in Chinese]. Chin. J. Bioinform. 18, 103–108 (2020).
Wrobel, A. G. et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 27, 763–767 (2020).
Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293–299 (2021).
Zhou, P. et al. Addendum: A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 588, E6 (2020).
Boni, M. F. et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417(2020).
Lam, T. T. et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285 (2020).
Xiao, K. et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289(2020).
Li, X. et al. Pathogenicity, tissue tropism and potential vertical transmission of SARSr-CoV-2 in Malayan pangolins. Preprint at https://doi.org/10.1101/2020.06.22.164442 (2020).
Zhou, H. et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 30, 2196–2203 (2020).
Zhou, H. et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184, 4380–4391 (2021).
Murakami, S. et al. Detection and characterization of bat sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 26, 3025–3029 (2020).
Delaune, D. et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 12, 6563 (2021).
Wacharapluesadee, S. et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 12, 972 (2021).
Temmam, S. et al. Coronaviruses with a SARS-CoV-2-like receptor-binding domain allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. Preprint at https://doi.org/10.21203/rs.3.rs-871965/v1 (2021).
Bouvet, M. et al. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl Acad. Sci. USA 109, 9372–9377 (2012).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 19, 9–20 (2018).
Grubaugh, N. D. et al. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 4, 10–19 (2019).
Rockett, R. J. et al. Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 26, 1398–1404 (2020).
Popa, A. et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 12, eabe2555 (2020).
Oude Munnink, B. B. et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 26, 1405–1410 (2020).
Miller, D. et al. Full genome viral sequences inform patterns of SARS-CoV-2 spread into and within Israel. Nat. Commun. 11, 5518 (2020).
du Plessis, L. et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 371, 708–712 (2021). This milestone paper describes the genomic surveillance of SARS-CoV-2 in the UK and used an analysis of more than 50,000 genome sequences to analyse the structure of SARS-CoV-2 lineages at a fine scale.
Zhong, N. S. et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 362, 1353–1358 (2003).
Lu, J. et al. Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 181, 997–1003 (2020). A detailed description of the genomic epidemiology of SARS-CoV-2 in Guangdong province, China, illustrating how genomic surveillance facilitated outbreak containment in China.
Liu, Z. et al. Identification of common deletions in the spike protein of severe acute respiratory syndrome coronavirus 2. J. Virol. 94, e00790-20 (2020).
Imported coronavirus variant case reported in Guangdong. XINHUA http://www.xinhuanet.com/english/2021-01/03/c_139637931.htm (3 January 2021).
Guangdong reports imported coronavirus variant case. XINHUA http://www.xinhuanet.com/english/2021-01/06/c_139646690.htm (6 January 2021).
Holshue, M. L. et al. First case of 2019 novel coronavirus in the United States. New Engl. J. Med. 382, 929–936 (2020).
World Health Organization. Coronavirus disease 2019 (COVID-19) Situation Report – 26 https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200215-sitrep-26-covid-19.pdf?sfvrsn=a4cc6787_2 (WHO, 2020).
Worobey, M. et al. The emergence of SARS-CoV-2 in Europe and North America. Science 370, 564–570 (2020).
Gonzalez-Reiche, A. S. et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 369, 297–301 (2020). This paper analysed the early transmission dynamics of SARS-CoV-2 in New York City, highlighting viral introductions from Europe to the USA.
Deng, X. et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 369, 582–587 (2020).
Bedford, T. et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 370, 571–575 (2020). This paper describes the cryptic transmission of SARS-CoV-2 in the USA.
Maurano, M. T. et al. Sequencing identifies multiple early introductions of SARS-CoV-2 to the New York City region. Genome Res. 30, 1781–1788 (2020).
Fauver, J. R. et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 181, 990–996 (2020). This study describes the long-distance domestic spread of SARS-CoV-2 in the USA.
Moreno, G. K. et al. Revealing fine-scale spatiotemporal differences in SARS-CoV-2 introduction and spread. Nat. Commun. 11, 5558 (2020).
Duchene, S. et al. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 6, veaa061 (2020).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Di Giorgio, S., Martignano, F., Torcia, M. G., Mattiuz, G. & Conticello, S. G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 6, eabb5813 (2020).
Simmonds, P. Rampant C→U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere 5, e00408-20 (2020).
Lu, G., Wang, Q. & Gao, G. F. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 23, 468–478 (2015).
Wang, Q., Wong, G., Lu, G., Yan, J. & Gao, G. F. MERS-CoV spike protein: targets for vaccines and therapeutics. Antiviral Res. 133, 165–177 (2016).
Korber, B. et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020). An important paper that provided the initial evidence that the D614G substitution increased the infectivity—and therefore transmissibility—of SARS-CoV-2.
Volz, E. et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell 184, 64–75 (2021).
Plante, J. A. et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592, 116–121 (2020).
Zhang, L. et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 11, 6013 (2020).
Yurkovetskiy, L. et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 183, 739–751 (2020).
Hou, Y. J. et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 370, 1464–1468 (2020).
Lassaunière, R. et al. SARS-CoV-2 Spike Mutations Arising in Danish Mink and their Spread to Humans (Working paper of SSI), https://files.ssi.dk/Mink-cluster-5-short-report_AFO2 (2020).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57 (2021).
Volz, E. et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 593, 266–269 (2021).
Davies, N. G. et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372, eabg3055 (2020).
Tegally, H. et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARSCoV-2) lineage with multiple spike mutations in South Africa. Preprint at https://doi.org/10.1101/2020.12.21.20248640 (2020).
Faria, N. R. et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (2021).
Naveca, F. et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. Virological https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spikeprotein/585 (2021).
O’Toole, Á. & Hill, V. SARS-CoV-2 lineages: B.1.617.2 report. https://cov-lineages.org/global_report_B.1.617.2.html (2021).
MacLean, O. A. et al. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 19, e3001115 (2021).
Martin, D. P. et al. The emergence and ongoing convergent evolution of the N501Y lineages coincides with a major global shift in the SARS-CoV-2 selective landscape. Preprint at https://doi.org/10.1101/2021.02.23.21252268 (2021).
Kupferschmidt, K. Viral evolution may herald new pandemic phase. Science 371, 108–109 (2021).
Wang, P. et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 593, 130–135 (2021).
Liu, C. et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 184, 4220–4236 (2021).
Liu, J. et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature 596, 273–275 (2021).
Collier, D. A. et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593, 136–141 (2021).
Nelson, G. et al. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. Preprint at https://doi.org/10.1101/2021.01.13.426558 (2021).
Wibmer, C. K. et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 27, 622–625 (2021).
Garcia-Beltran, W. F. et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 184, 2372–2383 (2021).
Dejnirattisai, W. et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 184, 2939–2954 (2021).
Su, Y. C. F. et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS-CoV-2. mBio 11, e01610-20 (2020).
Young, B. E. et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet 396, 603–611 (2020).
Gong, Y. N. et al. SARS-CoV-2 genomic surveillance in Taiwan revealed novel ORF8-deletion mutant and clade possibly associated with infections in Middle East. Emerg. Microbes Infect. 9, 1457–1466 (2020).
Du, P. et al. Genomic surveillance of COVID-19 cases in Beijing. Nat. Commun. 11, 5503 (2020).
Pan, A. et al. Association of public health interventions with the epidemiology of the COVID-19 outbreak in Wuhan, China. J. Am. Med. Assoc. 323, 1915–1923 (2020).
Hao, X. et al. Reconstruction of the full transmission dynamics of COVID-19 in Wuhan. Nature 584, 420–424 (2020). A comprehensive study of the transmission dynamics of COVID-19 in Wuhan, China, through time, providing important lessons learnt from the interventions in the city.
Xu, X. et al. Seroprevalence of immunoglobulin M and G antibodies against SARS-CoV-2 in China. Nat. Med. 26, 1193–1195 (2020).
Liu, A. et al. Seropositive prevalence of antibodies against SARS-CoV-2 in Wuhan, China. JAMA Netw. Open. 3, e2025717 (2020).
Chinese Center for Disease Control and Prevention. Scientific Understanding of the Prevalence of SARS-CoV-2: Q&A on the Results of National COVID-19 Seroepidemiological Survey in China, http://www.chinacdc.cn/yw_9324/202012/t20201228_223494.html (Chinese CDC, 2020).
Li, Z. et al. Antibody seroprevalence in the epicenter Wuhan, Hubei, and six selected provinces after containment of the first epidemic wave of COVID-19 in China.Lancet Reg. Health West Pac. 8, 100094 (2021).
Li, R. et al. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 368, 489–493 (2020).
Niehus, R., De Salazar, P. M., Taylor, A. R. & Lipsitch, M. Using observational data to quantify bias of traveller-derived COVID-19 prevalence estimates in Wuhan, China. Lancet Infect. Dis. 20, 803–808 (2020).
Cao, S. Y. et al. Post-lockdown SARS-CoV-2 nucleic acid screening in nearly ten million residents of Wuhan, China. Nat. Commun. 11,5917 (2020).
Jia, J. S. et al. Population flow drives spatio-temporal distribution of COVID-19 in China. Nature 582, 389–394 (2020).
Wu, J. T., Leung, K. & Leung, G. M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet 395, 689–697 (2020).
Lai, S. et al. Effect of non-pharmaceutical interventions to contain COVID-19 in China. Nature 585, 410–413 (2020). This study quantified the effects of various non-pharmaceutical interventions and their timings on COVID-19, providing early evidence that informed response efforts around the world.
Chinazzi, M. et al. The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak. Science 368, 395–400 (2020).
Zhang, J. et al. Evolving epidemiology and transmission dynamics of coronavirus disease 2019 outside Hubei province, China: a descriptive and modelling study. Lancet Infect. Dis. 20, 793–802 (2020).
Kraemer, M. U. G. et al. The effect of human mobility and control measures on the COVID-19 epidemic in China. Science 368, 493–497 (2020).
Leung, K., Wu, J. T., Liu, D. & Leung, G. M. First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: a modelling impact assessment. Lancet 395, 1382–1393 (2020).
Zhang, Z.-B. et al. Countries of origin of imported COVID-19 cases into China and measures to prevent onward transmission. J. Travel Med. 27, taaa139 (2020).
Bai, L. et al. Controlling COVID-19 transmission due to contaminated imported frozen food and food packaging. China CDC Wkly 3, 30–33 (2021).
Pang, X. et al. Cold-chain food contamination as the possible origin of COVID-19 resurgence in Beijing. Natl Sci. Rev. 7, 1861–1864 (2020).
Xing, Y., Wong, G. W. K., Ni, W., Hu, X. & Xing, Q. Rapid response to an outbreak in Qingdao, China. N. Engl. J. Med. 383, e129 (2020).
Bogoch, I. I. et al. Potential for global spread of a novel coronavirus from China. J. Travel Med. 27, taaa0111 (2020).
Lai, S., Bogoch, I. I., Watts, A., Khan, K. & Tatem, A. Preliminary risk analysis of 2019 novel coronavirus spread within and beyond China. WorldPop https://www.worldpop.org/events/china (2020).
Yang, J. et al. Uncovering two phases of early intercontinental COVID-19 transmission dynamics. J. Travel Med. 27, taaa200 (2020).
Pullano, G. et al. Novel coronavirus (2019-nCoV) early-stage importation risk to Europe, January 2020. Euro Surveill. 25, 2000057 (2020).
Tian, H. et al. An investigation of transmission control measures during the first 50 days of the COVID-19 epidemic in China. Science 368, 638–642 (2020).
Wells, C. R. et al. Impact of international travel and border control measures on the global spread of the novel 2019 coronavirus outbreak. Proc. Natl Acad. Sci. USA 117, 7504–7509 (2020).
Russell, T. W. et al. Effect of internationally imported cases on internal spread of COVID-19: a mathematical modelling study. Lancet Public Health 6, e12–e20 (2021).
Devi, S. COVID-19 resurgence in Iran. Lancet 395, 1896 (2020).
Giordano, G. et al. Modelling the COVID-19 epidemic and implementation of population-wide interventions in Italy. Nat. Med. 26, 855–860 (2020).
Salje, H. et al. Estimating the burden of SARS-CoV-2 in France. Science 369, 208–211 (2020).
Flaxman, S. et al. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 584, 257–261 (2020).
Bushman, D. et al. Detection and genetic characterization of community-based SARS-CoV-2 infections — New York City, March 2020. MMWR Morb. Mortal. Wkly Rep. 69, 918–922 (2020).
Rossen, L. M., Branum, A. M., Ahmad, F. B., Sutton, P. & Anderson, R. N. Excess deaths associated with COVID-19, by age and race and ethnicity — United States, January 26–October 3, 2020. MMWR Morb. Mortal. Wkly Rep. 69, 1522–1527 (2020).
Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369, 1255–1260 (2020).
Silveira, M. F. et al. Population-based surveys of antibodies against SARS-CoV-2 in Southern Brazil. Nat. Med. 26, 1196–1199 (2020).
Acharya, R. & Porwal, A. A vulnerability index for the management of and response to the COVID-19 epidemic in India: an ecological study. Lancet Glob. Health 8, e1142–e1151 (2020).
Laxminarayan, R. et al. Epidemiology and transmission dynamics of COVID-19 in two Indian states. Science 370, 691–697 (2020).
Massinga Loembé, M. et al. COVID-19 in Africa: the spread and response. Nat. Med. 26, 999–1003 (2020).
Gilbert, M. et al. Preparedness and vulnerability of African countries against importations of COVID-19: a modelling study. Lancet 395, 871–877 (2020).
Pullano, G. et al. Underdetection of cases of COVID-19 in France threatens epidemic control. Nature 590, 134–139 (2021).
Rice, B. L. et al. Variation in SARS-CoV-2 outbreaks across sub-Saharan Africa. Nat. Med. 27, 447–453 (2021).
Salyer, S. J. et al. The first and second waves of the COVID-19 pandemic in Africa: a cross-sectional study. Lancet 397, 1265–1275 (2021).
Pei, S., Kandula, S. & Shaman, J. Differential effects of intervention timing on COVID-19 spread in the United States. Sci. Adv. 6, eabd6370 (2020).
Brauner, J. M. et al. Inferring the effectiveness of government interventions against COVID-19. Science 371, eabd9338 (2020). A chronological and global dataset was constructed to compare the effectiveness of different NPIs in reducing COVID-19 transmission among countries during the first wave of the COVID-19 pandemic.
Haug, N. et al. Ranking the effectiveness of worldwide COVID-19 government interventions. Nat. Hum. Behav. 4, 1303–1312 (2020).
Dehning, J. et al. Inferring change points in the spread of COVID-19 reveals the effectiveness of interventions. Science 369, eabb9789 (2020).
Baker, R. E., Yang, W. C., Vecchi, G. A., Metcalf, C. J. E. & Grenfell, B. T. Susceptible supply limits the role of climate in the early SARS-CoV-2 pandemic. Science 369, 315–319 (2020).
Han, E. et al. Lessons learnt from easing COVID-19 restrictions: an analysis of countries and regions in Asia Pacific and Europe. Lancet 396, 1525–1534 (2020).
Badr, H. S. et al. Association between mobility patterns and COVID-19 transmission in the USA: a mathematical modelling study. Lancet Infect. Dis. 20, 1247–1254 (2020).
Perkins, T. A. et al. Estimating unobserved SARS-CoV-2 infections in the United States. Proc. Natl Acad. Sci. USA 117, 22597–22602 (2020).
Centers for Disease Control and Prevention. COVID Data Tracker. https://covid.cdc.gov/covid-data-tracker/#cases_casesper100klast7days (US CDC, 2020).
The Ministry of Health, Brazil. Coronavirus, Brazil. https://covid.saude.gov.br/ (2021).
Lemey, P. et al. Untangling introductions and persistence in COVID-19 resurgence in Europe. Nature 595, 713–717 (2021).
Hodcroft, E. B. et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 595, 707–712 (2021).
European Centre for Disease Prevention and Control. COVID-19. https://www.ecdc.europa.eu/en/covid-19-pandemic (ECDC, 2021).
Kupferschmidt, K. & Wadman, M. Delta variant triggers new phase in the pandemic. Science 372, 1375–1376 (2021).
World Health Organization. Weekly epidemiological update on COVID-19 – 29 June 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---29-june-2021 (WHO, 2021).
WHO. Tracking SARS-CoV-2 variants, https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (2021).
Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (US CDC, 2021).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Li, Q. et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182, 1284–1294 (2020).
Greaney, A. J. et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29, 463–476 (2021).
Saad-Roy, C. M. et al. Epidemiological and evolutionary considerations of SARS-CoV-2 vaccine dosing regimes. Science 372, 363–370 (2021).
Kemp, S. A. et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 592, 277–282 (2021).
Cobey, S., Larremore, D. B., Grad, Y. H. & Lipsitch, M. Concerns about SARS-CoV-2 evolution should not hold back efforts to expand vaccination. Nat. Rev. Immunol. 21, 330–335 (2021).
Hanage, W. P. & Russell, C. A. Partial immunity and SARS-CoV-2 mutations. Science 372, 354 (2021).
Murray, C. J. L. & Piot, P. The potential future of the COVID-19 pandemic: will SARS-CoV-2 become a recurrent seasonal infection? J. Am. Med. Assoc. 325, 1249–1250 (2021).
Phillips, N. The coronavirus is here to stay—here’s what that means. Nature 590, 382–384 (2021).
Avanzato, V. A. et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell 183, 1901–1912 (2020).
Aydillo, T. et al. Shedding of viable SARS-CoV-2 after immunosuppressive therapy for cancer. N. Engl. J. Med. 383, 2586–2588 (2020).
Lawton, G. Exclusive: Two variants have merged into heavily mutated coronavirus. New Scientist https://www.newscientist.com/article/2268014-exclusive-two-variants-have-merged-into-heavily-mutated-coronavirus/ (16 February 2021).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
GISAID. UK reports New Variant, termed VUI 202012/01. https://www.gisaid.org/references/gisaid-in-the-news/uk-reports-new-variant-termed-vui-20201201/ (2021).
Song, S. et al. The global landscape of SARS-CoV-2 genomes, variants, and haplotypes in 2019nCoVR. Genom. Proteom. Bioinform. 18, 749–759 (2020).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Davies, N. G. et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 593, 270–274 (2021).
Frampton, D. et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: a whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 21, 1246–1256 (2021).
Graham, M. S. et al. Changes in symptomatology, reinfection, and transmissibility associated with the SARS-CoV-2 variant B.1.1.7: an ecological study. Lancet Public Health 6, e335–e345 (2021).
World Health Organization. Weekly epidemiological update on COVID-19 – 10 August 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---10-august-2021 (WHO, 2021).
European Centre for Disease Prevention and Control. Emergence of SARS-CoV-2 B.1.617 variants in India and situation in the EU/EEA. https://www.ecdc.europa.eu/en/publications-data/threat-assessment-emergence-sars-cov-2-b1617-variants (ECDC, 2021).
Delta Plus: Key things to know about new coronavirus variant. The Economic Times https://economictimes.indiatimes.com/news/et-explains/delta-plus-key-things-to-know-about-new-coronavirus-variant-in-india/articleshow/83739996.cms (24 June 2021).
Gu, H. et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369, 1603–1607 (2020).
Acknowledgements
We acknowledge the efforts of the World Health Organization in sharing the COVID-19 Weekly Epidemiological Update, the researchers that are part of the cov-lineages.org team (https://cov-lineages.org/) in assembling the records for new strains, the Johns Hopkins University Center for Systems Science and Engineering (JHU CSSE) for collating the COVID-19 case data (github.com/CSSEGISandData/COVID-19), and Google for producing and sharing the COVID-19 Community Mobility Reports (www.google.com/covid19/mobility/). We thank the researchers who generated and shared the sequencing data from the GISAID and NCBI GenBank databases; T. Hu, P. Wang, X. Yao and H. Song for helping to produce the figures; and E. C. Holmes, A. Tatem and J. Floyd for commenting on the manuscript. This work was supported by Key Research and Development Project of Shandong province (grant nos. 2020SFXGFY01 and 2020SFXGFY08), the National Key Research and Development Programme of China (grant no. 2020YFC0840800), the National Science and Technology Major Project (Grant no. 2018ZX10101004-002 and 2016ZX10004222-009), the National Natural Science Fund of China (81773498), the Academic Promotion Programme of Shandong First Medical University (2019QL006), and the Bill & Melinda Gates Foundation (INV-024911). W.S. was supported by the Taishan Scholars Program of Shandong Province (ts201511056).
Author information
Authors and Affiliations
Contributions
W.S. conceived the study. J.L. performed phylogenetic analysis and homology modelling. S.L. conducted the literature review on the global spread of SARS-CoV-2 and VOCs, and collected, analysed and visualized the data of the case numbers, VOC reports and human mobility, using publicly available data resources. W.S., J.L. and S.L. wrote the first draft of the manuscript. W.S. and G.F.G. proofread the manuscript and the pre-submission inquiry.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Malik Peiris and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 The date of the earliest sequence of VOC detected in each country, territory or area.
a, VOC Alpha (lineage B.1.1.7). b, VOC Beta (lineage B.1.351). c, VOC Gamma (lineage P.1). d, VOC Delta (B.1.617.2). Reporting date of the earliest sequence of each VOC used records published at https://cov-lineages.org/, as of 30 July 2021, derived from publicly available sequence data in GISAID, shared by international sequencing efforts. The areas without data are shown in grey.
Supplementary information
Supplementary Table 1
The top 50 ranked countries with the highest number of cases of COVID-19 reported as of 8 August 2021 and the available Google mobility data.
Rights and permissions
About this article

Cite this article
Li, J., Lai, S., Gao, G.F. et al. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 600, 408–418 (2021). https://doi.org/10.1038/s41586-021-04188-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-04188-6
This article is cited by
-
Association of dietary inflammatory index and the SARS-CoV-2 infection incidence, severity and mortality of COVID-19: a systematic review and dose-response meta-analysis
Nutrition Journal (2024)
-
Precision nutrition to reset virus-induced human metabolic reprogramming and dysregulation (HMRD) in long-COVID
npj Science of Food (2024)
-
LAPIS is a fast web API for massive open virus sequencing data
BMC Bioinformatics (2023)
-
Binding and inactivation of human coronaviruses, including SARS-CoV-2, onto purified clinoptilolite-tuff
Scientific Reports (2023)
-
Therapeutic strategies for COVID-19: progress and lessons learned
Nature Reviews Drug Discovery (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
