
Abstract
Single-particle electron cryo-microscopy (cryo-EM) is a powerful method for solving the three-dimensional structures of biological macromolecules. The technological development of transmission electron microscopes, detectors and automated procedures in combination with user-friendly image processing software and ever-increasing computational power have made cryo-EM a successful and expanding technology over the past decade1. At resolutions better than 4 Å, atomic model building starts to become possible, but the direct visualization of true atomic positions in protein structure determination requires much higher (better than 1.5 Å) resolution, which so far has not been attained by cryo-EM. The direct visualization of atom positions is essential for understanding the mechanisms of protein-catalysed chemical reactions, and for studying how drugs bind to and interfere with the function of proteins2. Here we report a 1.25 Å-resolution structure of apoferritin obtained by cryo-EM with a newly developed electron microscope that provides, to our knowledge, unprecedented structural detail. Our apoferritin structure has almost twice the 3D information content of the current world record reconstruction (at 1.54 Å resolution3). We can visualize individual atoms in a protein, see density for hydrogen atoms and image single-atom chemical modifications. Beyond the nominal improvement in resolution, we also achieve a substantial improvement in the quality of the cryo-EM density map, which is highly relevant for using cryo-EM in structure-based drug design.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
The atomic models have been deposited in the Protein Data Bank (PDB) with the following accession codes: 1.56 Å structure (COW) 6Z9F, 1.55 Å structure 6Z9E, 1.33 Å structure 7A6B, 1.25 Å structure 6Z6U, and 1.15 Å structure 7A6A. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank as follows: 1.56 Å map (COW, EMD-11122), 1.55 Å map (EMD-11121), 1.33 Å map (EMD-11669), 1.25 Å map (EMD-11103) and 1.15 Å map (EMD-11668).
References
Kühlbrandt, W. The resolution revolution. Science 343, 1443–1444 (2014).
Lüdtke, S. et al. Sub-ångström-resolution crystallography reveals physical distortions that enhance reactivity of a covalent enzymatic intermediate. Nat. Chem. 5, 762–767 (2013).
Kato, T. et al. The 1.54 Å resolution structure of apoferritin by CRYOARM300 with Cold-FEG. Microsc. Microanal. 25, 998–999 (2019).
Hamaguchi, T. et al. A new cryo-EM system for single particle analysis. J. Struct. Biol. 207, 40–48 (2019).
Tiemeijer, P. C., Bischoff, M., Freitag, B. & Kisielowski, C. Using a monochromator to improve the resolution in TEM to below 0.5Å. Part I: Creating highly coherent monochromated illumination. Ultramicroscopy 114, 72–81 (2012).
Müller, H. et al. Aplanatic imaging systems for the transmission electron microscope. Nucl. Instrum. Meth. A 645, 20–27 (2011).
Stark, H. & Chari, A. Sample preparation of biological macromolecular assemblies for the determination of high-resolution structures by cryo-electron microscopy. Microscopy (Oxf.) 65, 23–34 (2016).
Karuppasamy, M., Karimi Nejadasl, F., Vulovic, M., Koster, A. J. & Ravelli, R. B. Radiation damage in single-particle cryo-electron microscopy: effects of dose and dose rate. J. Synchrotron Radiat. 18, 398–412 (2011).
Frank, J. Generalized single-particle cryo-EM-a historical perspective. Microscopy (Oxf.) 65, 3–8 (2016).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Bracewell, B. L. & Veigele, W. J. in Advances in X-Ray Analysis Vol. 15 (eds Heinrich, K. F. J. et al.) 352–364 (Springer US, 1972).
Williams, J. F. Electron scattering from atomic hydrogen. III. Absolute differential cross sections for elastic scattering of electrons of energies from 20 to 680 eV. J. Phys. B 8, 2191–2199 (1975).
Carter, C., March, N. H. & Vincent, D. X-ray and electron scattering by molecular hydrogen. Proc. Phys. Soc. 71, 2–16 (1958).
Harauz, G. & van Heel, M. Exact filters for general geometry three dimensional reconstruction. Optik (Stuttg.) 73, 146–156 (1986).
Grigorieff, N. Resolution measurement in structures derived from single particles. Acta Crystallogr. D 56, 1270–1277 (2000).
Heinrich, D., Diederichsen, U. & Rudolph, M. G. Lys314 is a nucleophile in non-classical reactions of orotidine-5′-monophosphate decarboxylase. Chemistry 15, 6619–6625 (2009).
Homans, S. W. Water, water everywhere—except where it matters? Drug Discov. Today 12, 534–539 (2007).
Schiebel, J. et al. Intriguing role of water in protein-ligand binding studied by neutron crystallography on trypsin complexes. Nat. Commun. 9, 3559 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Bromberg, R., Guo, Y., Borek, D. & Otwinowski, Z. High-resolution cryo-EM reconstructions in the presence of substantial aberrations. IUCrJ 7, 445–452 (2020).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Wu, M., Lander, G. C. & Herzik, M. A., Jr. Sub-2 Angstrom resolution structure determination using single-particle cryo-EM at 200 keV. J. Struct. Biol. X 4, 100020 (2020).
Russo, C. J. & Henderson, R. Ewald sphere correction using a single side-band image processing algorithm. Ultramicroscopy 187, 26–33 (2018).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D 66, 22–25 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011).
Singh, K. et al. Discovery of a regulatory subunit of the yeast fatty acid synthase. Cell 180, 1130–1143.e1120 (2020).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Acknowledgements
We thank J. E. Schliep for his contribution in the early phase of the project; M. Lüttich for setting up the computational facilities; and M. Link and S. Dalaikhuu for their technical contribution in the initial setup of the microscope. This work was supported by the Max Planck Society and the Deutsche Forschungsgemeinschaft (grants SFB 860 to H.S.).
Author information
Authors and Affiliations
Contributions
H.S., K.M.Y. and N.F. set up the Krios Mono/BCOR microscope. E.P. and A.C. developed the purification strategy and E.P. purified the complex. K.M.Y. prepared grids and collected EM data. K.M.Y. and N.F. did the cryo-EM image processing analysis. A.C. built and refined the atomic models. The manuscript was written by H.S. with input from all authors. H.S. initiated and orchestrated the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Henning Stahlberg, Serban Ilca and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
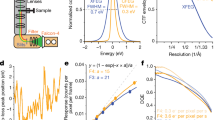
Extended Data Fig. 1 Cryo-EM structure determination.
a, Image processing pipeline. See Methods for details. b, Fourier-shell-correlation plots for independently refined half-maps (Half maps) and full map versus model (Map to model). c, Histogram of local resolution for the final 1.25 Å map obtained with relion postprocess using a small soft spherical mask. d, Final map colored by local resolution. e, Defocus distribution for the total set of 10,398 micrographs. f, Linear magnification distortions as estimated by relion_ctf_refine for the present Krios Mono/BCOR data and data from a standard Titan Krios (EMPIAR-10216). g, Relative magnification variation in the present data as determined by relion_ctf_refine. h, Angular distribution for the final map.
Extended Data Fig. 2 Low-dose cryo-EM micrographs of human apoferritin.
Exemplary micrographs (left) acquired with a total dose of ~50e-Å2 are shown with their power spectra (PS), equi-phase average (EPA) and the fit of the contrast transfer function (Fit). A total number of 10.398 similar micrographs has been recorded. Numbers indicate the respective defocus in μm and the maximum resolution (in Å) in the power spectra as estimated by Gctf22. White arrow heads denote the transition from areas with particles in dense packing to areas devoid of particles indicating a very thin layer of vitrified ice, as required for high-resolution imaging.
Extended Data Fig. 3 Comparing high-resolution features obtained by X-ray crystallography and cryo-EM.
For the comparison, we selected the same tyrosine residue (Tyr32 in human apoferritin, Tyr34 in frog) from our structures obtained at different resolutions with published crystallographic data of apoferritin at the indicated resolution. The same residue is shown at high (red) and low (grey) threshold to better judge the structural details and the map quality. Density that can be attributed to hydrogen atoms can barely be seen in any of the X-ray structures. Even at 1.06 Å resolution only weak density for hydrogens can be detected while the cryo-EM reconstruction already reveals some density for hydrogens at 1.35 Å resolution. At high thresholds, the separation into clearly distinct atoms can only be seen in the 1.06 Å resolution X-ray map but not in lower resolution X-ray data. In case of our cryo-EM reconstructions we can see individual atoms starting at 1.35 Å resolution or better. Cryo-EM structures at 1 Å resolution are currently not possible because they would require unrealistically high particle number statistics even with the Krios Mono/BCOR microscope (see Fig. 2).
Extended Data Fig. 4 Structural features of our cryo-EM maps at 1.55/1.25 Å resolution compared to the thus far reported highest-resolution map at 1.54 Å resolution (EMDB-9865).
Three apoferritin residues (His151, Phe51, Arg76) are shown at two different density thresholds in three cryo-EM maps. Row one depicts our present high-resolution map at 1.25 Å; resolution and row three shows a structure at 1.55 Å; resolution obtained from a smaller subset of the same data. Only 22.000 particles were necessary for this reconstruction to obtain 1.55 Å resolution which is a 5.5× lower particle statistics compared to the 1.54 Å Jeol CryoARM300 map (second row). The low-threshold density meshes are always shown in grey and H atoms (white sticks) are included in the corresponding atomic models. Only in the Krios Mono/BCOR structure at 1.25 Å; resolution density becomes visible to accommodate all hydrogen atoms. At higher thresholds the two structures at 1.54 Å and 1.55 Å resolution nicely maintain the shapes of the sidechains but only in the structure at 1.25 Å; resolution individual atoms become clearly separated from each other indicating true atomic resolution.
Extended Data Fig. 5 Visualization of a single atom chemical modification.
Cysteine 90 of human apoferritin is located at the surface of the macromolecular complex. The solvent-exposed regions are usually determined at lower local-resolution in cryo-EM but in our high-resolution structure it is still sufficient to visualize a single atom oxygen modification (marked by a square on the entire molecule and by the red circle in the close-up).
Extended Data Fig. 6 Relevant map-model building parameters and solvent molecules.
a, As expected for such high-resolution structures, the calculated model validation parameters are in a very good range for all cryo-EM map/models. The results from the three structures at 1.56/1.55/1.54 Å resolution can best be compared because they were determined at very similar resolution. When comparing those structures, all validation parameters are in favour of the Krios Mono/BCOR structures. Their atomic models have better geometries and more waters were built into the maps compared to the Jeol 1.54 Å map. b, The number of water molecules that we localized in a structure rises with resolution of a map, which is true for X-ray and for cryo-EM. The plot shows the number of water molecules found in the various X-ray structures (red dots) in relation to the cryo-EM maps (Relion-blue, COW-orange and Jeol-magenta). At the 1.5 Å level, substantially more water molecules were identified for both Krios Mono/BCOR reconstructions compared to the Jeol map at very similar nominal resolution. We obtained the highest number of waters for the 1.56 Å map that was calculated with the COW software. For this model, the number of built waters gets very close to the X-ray data at this resolution. It is noteworthy that the number of waters that were located in the X-ray structures is still generally higher than for the cryo-EM maps for reasons that are currently unknown.
Extended Data Fig. 7 Improving the image acquisition speed over the time of the project.
a, Several changes were made in the data acquisition scheme of the Titan Krios Mono/BCOR microscope. We started with manual data collection and then implemented several different EPU versions to increase the speed. EPU#1 was similar in speed to manual acquisition with one image per hole. EPU#2 required the automation of the C3 aperture and allowed us to take three images per hole. Only with EPU#3 we had a version allowing for automated re-centering of the monochromator which reduced the number of discarded images due to large dose variations. The currently highest speed in data acquisition is EPU#4 for which we reach roughly 100 movies per hour using the recording scheme shown in b) (only available since Dec 2019). Most data of this project was recorded with the EPU#3 scheme. b, To reach an acquisition speed of 100 micrographs/hour we move the stage only once to record data from 9 ice holes on a custom made EM grid (Quantifoil R1.2/1.0). Image/beam shift is applied to reach all 45 acquisition areas. The BCOR is tuned to compensate for image shift induced coma. Using this scheme we would also benefit significantly from a faster camera like the Falcon 4 (Thermo Fischer Scientific). Just by replacing the camera this would allow us to reach an almost 5 times higher recording speed of 500 micrographs per hour (F4 in a).
Supplementary information
Video 1
This video shows ten amino acids of apoferritin at atomic resolution with the cryo-EM density at two different thresholds. (Software: Cinema4D-R14).
Rights and permissions
About this article

Cite this article
Yip, K.M., Fischer, N., Paknia, E. et al. Atomic-resolution protein structure determination by cryo-EM. Nature 587, 157–161 (2020). https://doi.org/10.1038/s41586-020-2833-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2833-4
This article is cited by
-
High-resolution cryo-EM of the human CDK-activating kinase for structure-based drug design
Nature Communications (2024)
-
Improving resolution and resolvability of single-particle cryoEM structures using Gaussian mixture models
Nature Methods (2024)
-
A continuum of amorphous ices between low-density and high-density amorphous ice
Communications Chemistry (2024)
-
Anaerobic cryoEM protocols for air-sensitive nitrogenase proteins
Nature Protocols (2024)
-
Metallo-supramolecular branched polymer protects particles from air-water interface in single-particle cryo-electron microscopy
Communications Biology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
