
Abstract
Research into the immunological processes implicated in cancer has yielded a basis for the range of immunotherapies that are now considered the fourth pillar of cancer treatment (alongside surgery, radiotherapy and chemotherapy). For some aggressive cancers, such as advanced non-small-cell lung carcinoma, combination immunotherapies have resulted in unprecedented treatment efficacy for responding patients, and have become frontline therapies. Individualized immunotherapy, enabled by the identification of patient-specific mutations, neoantigens and biomarkers, and facilitated by advances in genomics and proteomics, promises to broaden the responder patient population. In this Perspective, we give an overview of immunotherapies leveraging engineering approaches, including the design of biomaterials, delivery strategies and nanotechnology solutions, for the realization of individualized cancer treatments such as nanoparticle vaccines customized with neoantigens, cell therapies based on patient-derived dendritic cells and T cells, and combinations of theranostic strategies. Developments in precision cancer immunotherapy will increasingly rely on the adoption of engineering principles.
Similar content being viewed by others

Main
Cancer immunotherapies—which harness and boost the body’s immune system to target and kill tumour cells—include antibodies that block suppressive immune-checkpoint pathways, cellular therapies based on dendritic cells (DCs) and engineered T cells, and vaccines that trigger antigen-specific immune responses in tumours. Blocking antibodies specific for the immune checkpoint proteins cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death receptor-1 (PD-1) have been game-changers in clinical cancer therapy1,2,3,4,5. These antibodies, designed to liberate T cells from the immunosuppression mediated by the CTLA-4 and PD-1 pathways, promote potent and durable T-cell responses that can eliminate tumours and lead to cancer remission3,6. Still, only 10–30% of patients benefit from such immune-checkpoint blockade3,6, and the co-administration of both anti-CTLA-4 and anti-PD-1 antibodies for synergistic tumour killing can lead to serious immune-related toxicities. For instance, one clinical study of patients treated with dual anti-CTLA-4 and anti-PD-1 immunotherapy reported that 53% of those patients experienced grade-3 or grade-4 adverse events, including hepatic, gastrointestinal and renal disorders7. There is thus strong interest in improving patient response rates and the safety of cancer immunotherapies.
One strategy for achieving this objective would be to combine immune-checkpoint blockade with cellular therapies or therapeutic vaccines8,9,10,11,12,13,14,15,16,17. Cellular therapies based on patient-derived DCs (obtained from the ex vivo differentiation of peripheral blood monocytes) loaded with tumour-associated antigens (TAAs) can be infused back into the patient to enhance T-cell activation and tumour-cell killing18,19. Similarly, T cells isolated from a patient’s blood can be purified to contain particular T-cell populations that can be genetically modified to promote anti-tumour efficacy. Unfortunately, the production of TAA-presenting DCs, or of tumour-specific T cells, is labour-intensive and is associated with variable yields and quality. In light of these limitations, acellular cancer vaccines and combination immunotherapies may have some advantages.
Recent advances in genomics and proteomics focussed on the tumour mutanome have revealed that every tumour has a unique set of ‘driver’ mutations and ‘passenger’ mutations20,21,22. This observation has provided unique opportunities for personalized therapies. Tumour cells expressing mutated proteins (neoantigens) present these new epitopes in the context of major histocompatibility complex (MHC) molecules. In contrast to TAAs, whose expression is shared among healthy and tumour cells, neoantigens arise from mutations in tumours and are, therefore, fully restricted to tumour cells. Thus, immunotherapies that capitalize on rich genomic and proteomic data to develop personalized strategies based on neoantigens enable the highly specific targeting of tumour cells without risking healthy tissues and without being limited by immune tolerance mechanisms.
The prospect of neoantigen-directed immunotherapies providing cancer treatments customized to individual patients has galvanized researchers working in cancer immunotherapy20,21,22. Yet, the workflow for generating neoantigen-targeted therapies is complex. Whole exome DNA and RNA sequencing of patient-derived tumour cells is followed by the application of computational tools for neoantigen identification (by taking into account factors such as predicted proteasome processing and MHC class-I and class-II binding affinities); the ‘hits’ can then be further narrowed down with mass-spectrometry analyses of immunoprecipitated peptides. Once the top neoantigen candidates are identified, they can be used to screen patient-derived samples for the presence of neoantigen-specific T cells. The concept of neoantigen-based personalized immunotherapy was just recently demonstrated in murine models of cancer23,24,25,26, but has already been translated to proof-of-concept phase-I clinical trials with small cohorts of patients with advanced melanoma27,28 or glioblastoma multiforme29,30.
In this Perspective, we highlight state-of-the-art engineering strategies for improving the efficacy and potency of cancer immunotherapy. We focus on recent advances in biomaterials design, drug-delivery strategies and nanotechnology that promise to accelerate progress in the development of patient-specific cancer immunotherapies (Fig. 1), including peptide-based vaccines featuring neoantigens, gene therapies designed to deliver neoantigens or immunomodulatory proteins, cellular therapies based on patient-derived DCs and T cells, and nanotechnology for image-guided theranostic applications. We argue that biomaterial-based drug-delivery strategies offer exciting opportunities for personalized immunotherapy and precision medicine. We also provide an overview of the advantages and disadvantages of combination therapies involving photothermal (PTT), photodynamic (PTD) and radiation therapies for advanced cancers (Table 1).

(1) Tumour samples from cancer patients are collected. (2) The genomic sequence of the tumour is compared with the somatic genome sequence so as to locate mutations, which are processed through multiple algorithms for the prediction of neoantigens. (3,4) Neoantigen-specific DNA, mRNA and peptides are generated (3) and formulated into a personalized nanomedicine for combination cancer immunotherapy (4). (5) Neoantigens can also be loaded on APCs to generate DC vaccines or neoantigen-specific T cells ex vivo. (6) Alternatively, genes encoding scFv or TCRs specific to neoantigens can be transduced into peripheral lymphocytes to generate tumour-reactive T cells for adoptive transfer into the patient. APC, antigen presenting cell. TCR, T-cell receptor.
Neoantigen peptide vaccines
A comprehensive study of vaccination using neoantigen peptides, reported in 2012 (ref. 23), led to the identification of over 900 non-synonymous point mutations in B16F10 murine melanoma cells. The list of mutated peptide sequences was processed through MHC-binding prediction tools, which yielded a shortlist of 50 peptides that were then selected for immunogenicity screening in mice. Subcutaneous injection of neoantigen or wild-type peptides, together with the adjuvant polyinosinic:polycytidylic acid (poly(I:C); a Toll-like receptor (TLR)-3 agonist), led to neoantigen-specific, interferon (IFN)-γ-associated T-cell responses to five of the 50 peptides. When tested in a therapeutic setting and compared with adjuvant alone or with the absence of treatment, neoantigen vaccination significantly slowed the growth of B16F10 tumours. In 2014, tumour exome sequencing was employed in conjunction with mass spectrometry to identify neoantigens in a hindlimb MC-38 tumour model24. Here, mice that were vaccinated intraperitoneally with long neoantigen peptides (25–30mer amino acids) together with anti-CD40 monoclonal antibody and poly(I:C) adjuvant showed a reduction in tumour growth and an increase in tumour-infiltrating neoantigen-specific CD8+ T cells. In 2017, first-in-man phase-I clinical trials of the treatment of advanced melanoma with neoantigen peptide vaccines following surgical resection of the tumour were reported (Fig. 2)27. Each of the six patients received seven doses of 20 different neoantigen peptides mixed with poly(I:C) stabilized with poly-L-lysine (poly-ICLC) adjuvant27; neoantigen vaccination induced CD4+ and CD8+ T cells specific for 58 (60%) and 15 (16%) of the 97 unique neoantigens identified across the six patients. Four patients showed no recurrence at 25 months after vaccination, and two patients exhibited complete tumour regression after co-treatment with the checkpoint-blockade agent anti-PD-1. Along with another phase-I trial of a neoantigen encoding a messenger RNA vaccine28, these trials showed that personalized neoantigen vaccination, especially in combination with immune-checkpoint blockade, can unleash the full cytotoxic potential of neoantigen-specific T cells to kill tumours with limited adverse effects and underscored the clinical applicability of personalized neoantigen vaccines as a therapeutic strategy for long-term protection against tumour relapse and metastases.

Matched tumour-cell and normal-cell DNA from peripheral blood mononuclear cells and resected tumours are compared by whole-exome sequencing to detect mutations. Candidate neoantigen peptides are selected, synthesized and used for therapeutic vaccination in corresponding patients with poly-ICLC adjuvant. HLA, human leukocyte antigen. Figure reproduced from ref. 27, Springer Nature Ltd.
Delivery of peptide vaccines
The tantalizing results of neoantigen vaccination galvanized researchers working in personalized immunotherapy. Yet, producing potent anti-tumour neoantigen therapies safely and effectively is challenging, because the amino acid composition of neoantigen peptides can have significant effects on their isoelectric properties, meaning that the administration of a cocktail of soluble peptides can lead to their precipitation, deposition in off-target tissues, or dissemination through the systemic circulation without preferential targeting to lymphoid tissues. Such barriers to vaccine delivery can result in only a minor fraction of the injected peptides reaching the target lymphoid tissues, reducing vaccine efficacy. Efficient delivery strategies are thus needed to enhance the transport of neoantigens and adjuvant molecules to lymph nodes. Several research groups have reported enhanced immunogenicity with peptide vaccines when including oil-based adjuvants, such as Montanide, to create water-in-oil formulations that form depots for the slow release of antigen peptides31,32; however, these formulations are often associated with adverse effects such as abscess formation and sustained inflammation at the injection site, leading to the sequestration and deletion of antigen-specific T cells33,34,35. Therefore, neoantigen vaccines should be designed to maximize antigen delivery to lymph nodes while considering the aqueous solubility and physicochemical properties of neoantigen peptides. Additionally, the co-localized delivery of antigens and adjuvant molecules to the same intracellular compartments (such as endosomes containing TLRs) within antigen-presenting cells (APCs) is needed to achieve robust T-cell responses36,37. An ideal vaccine for neoantigens should be versatile yet easy to manufacture, as personalized vaccine products require rigorous quality-control and reproducible production.
To meet these demands, nanovaccine formulations are gaining momentum38. Nanoparticles with an optimal size for lymphatic trafficking (10–100 nm) are increasingly recognized as efficient carriers for the targeted delivery of antigens to APCs (refs. 39,40). For example, nanodiscs based on synthetic high-density lipoprotein (sHDL) can serve as an effective delivery system for therapeutic vaccination with neoantigen peptides (Fig. 3)16, owing to the nanodiscs’ small size (~10 nm in diameter), stability and biocompatibility. As large-scale manufacturability and clinical safety are major hurdles for the clinical translation of nanomedicines41, an additional advantage of the sHDL vaccine is that it builds on current good manufacturing practices (cGMPs) and clinical safety protocols previously demonstrated in clinical trials for cardiovascular applications42. Furthermore, sHDL nanodiscs can readily bind to lipoprotein cell receptors (such as scavenger receptor class B member 1 (SR-B1), which is overexpressed on APCs), therefore enhancing the targeted delivery of the nanodiscs to the relevant immune cells in lymph nodes43,44,45,46. Indeed, mice vaccinated with sHDL nanodiscs containing neoantigen peptides and the adjuvant CpG oligodeoxynucleotide elicited a ~47-fold higher frequency of neoantigen-specific CD8+ T cells when compared with the administration of soluble neoantigen peptide and CpG (ref. 16). Neoantigen nanodisc vaccination was also combined with immune-checkpoint blockade (using an anti-PD1 antibody) to achieve tumour eradication in >85% of mice bearing MC-38 colon carcinoma and B16F10 melanoma. A cocktail of nanodiscs carrying a TAA (namely, tyrosinase-related protein 2; TRP2) and two neoantigens had a noticeable advantage over vaccination with either TRP2 or two neoantigens alone, suggesting the potential advantages of vaccine formulations targeting a set of both TAAs and neoantigens16. Also of note, when compared with an intramuscular route, the subcutaneous administration of nanodiscs enhanced the delivery of antigens and adjuvants to lymph nodes, leading to an increase in the frequency of neoantigen-specific T cells and to the elimination of large established B16F10 tumours47,48. Whether sHDL vaccines personalized with neoantigens can achieve a significant survival benefit in the clinic remains to be seen16,49,50.

a, sHDL nanodiscs loaded with neoantigen peptides and adjuvants (such as CpG) are delivered to draining lymph nodes to induce immune activation signals 1 and 2. b, Neoantigen peptides can be identified via the DNA sequencing of tumour samples. Ag, antigen; TCR, T-cell receptor. Figure reproduced from ref. 16, Springer Nature Ltd.
The utilization of albumin—the most abundant protein in serum—as a vaccine-delivery carrier (‘albumin-hitchhiking’) offers an alternative approach to antigen and adjuvant delivery. This approach takes advantage of the biophysical properties, cellular interactions and molecular-transport mechanisms of serum albumin. The conjugation of TAAs and CpG to albumin-binding lipid tails enhanced T-cell responses by 30-fold and led to reduced tumour growth in both the TC-1 and B16F10 models when compared with free mixtures of TAAs and CpG (ref. 51). There are also related albumin-conjugate strategies for the delivery of chemotherapeutics and antigen–adjuvant combinations: an Evans blue analogue, AlbiVax, that binds to albumin for effective lymph-node draining has been tethered to antigens or to CpG to facilitate their systemic delivery (Fig. 4)52. AlbiVax enhanced the accumulation of a Cu64-labelled Adpgk neoantigen in lymph nodes by >40-fold when compared with soluble peptide vaccination in the MC-38 model, and vaccination with AlbiVax–Adpgk in the context of CpG adjuvant slowed tumour growth and protected animals against tumour rechallenge, an effect that could be potentiated by combination with checkpoint blockade. As albumin-bound drug conjugates have been approved by the United States Food and Drug Administration (FDA) for cancer treatment53, albumin-mediated vaccine formulations51,52,54,55 may have a simplified clinical-translation pathway for personalized-cancer-immunotherapy applications. Still, any autoimmunity against albumin or albumin-producing hepatocytes, as well as the potential for off-target toxicity of the albumin-hitchhiking therapeutics, should be carefully examined.

a, The molecular docking between albumin and a maleimide-functionalized Evans-blue derivative (MEB) leads to the formation of a nanocomplex. b, By conjugating the adjuvant CpG (AlbiCpG) or a tumour-specific antigen (AlbiAg) to MEB, CpG and Ag administered via the subcutaneous route are carried by the albumin–MEB complex to local draining lymph nodes (LN) and endocytosed by APCs. This is followed by maturation of APCs and enhanced antigen presentation, generating CD8+ T-cell responses. Figure reproduced from ref. 52, Springer Nature Ltd.
Self-assembled DNA–RNA nanocapsules are an alternative strategy for delivering neoantigen peptides. In a 2015 study54, DNA–RNA nanocapsules composed of CpG, Stat3 short-hairpin (sh)RNA (for reversing immunosuppression) and poly(ethylene glycol) (PEG)-grafted polypeptides for the delivery of neoantigens were shown to be internalized by bone-marrow-derived dendritic cells (BMDCs), and led to superior immune activation of the DCs when compared with CpG-containing larger parent particles or with CpG alone. In mice, DNA–RNA nanocapsules loaded with a MC-38 neoantigen peptide enhanced the neoantigen-specific CD8+ T-cell response and led to stronger anti-tumour effectiveness when compared to soluble peptide with CpG (ref. 56).
As an alternative approach, synthetic polymeric particles, such as poly 2-(hexamethyleneimino)ethyl methacrylate (PC7A) with an intrinsic ability to activate the stimulator of interferon genes (STING) pathway have also been employed for the induction of T-cell responses against neoantigens57. The small size of the PC7A nanoparticles (~29 nm) facilitated the effective lymphatic drainage of the antigen, as well as subsequent cellular uptake, cross-presentation and DC activation. The PC7A nanoparticles were efficacious in the B16F10 model of melanoma expressing the model antigen ovalbumin (OVA), and elicited potent anti-tumour responses when delivering a cocktail of TAAs and neoantigens in murine models of MC-38 colon carcinoma and B16F10 melanoma57.
There are also microscale delivery systems for vaccines. One example is the use of mixtures of mesoporous silica micro-rod scaffolds (MSRs), each separately adsorbed with CpG, granulocyte-macrophage colony-stimulating factor (GM-CSF) or polyethyleneimine (PEI), in complexation with antigens58. Including the PEI-adsorbed MSRs in the mixture significantly increased the expression of MHC-II and CD86 in BMDCs, as well as the in vitro production of interleukin (IL)-6 and of tumour necrosis factor (TNF)-α, potentially owing to lysosomal-destabilization and inflammasome activation mediated by PEI (ref. 59). Subcutaneous administration of MSR–PEI–OVA outperformed MSR–OVA in increasing antigen-specific CD8+ T cells, IFN-γ production, and the ratio of effector T cells and regulatory T (TREG) cells. Vaccination with mixtures of MSR–CpG, MSR–GM-CSF and MSR–PEI–neoantigens reduced the number of lung metastases in a mouse model of B16F10 and CT26 tumours, and exerted anti-tumour efficacy in synergy with anti-CTLA-4 checkpoint blockade58.
Taken together, these nanovaccines have yielded exciting proof-of-concept results for neoantigen-based personalized vaccination. It is notable that the performance of nanovaccines can be further improved when these are combined with immune-checkpoint-blockade therapy, highlighting the importance of addressing immunosuppression in the tumour microenvironment for effective cancer vaccination. Moreover, owing to improvements in the intracellular transport of cargo molecules and in the incorporation of targeting moieties, the biomaterials used for these nanovaccines showed few off-target toxicities and non-specific immune responses. On the way to making personalized nanovaccines a reality, remaining engineering challenges include how to control the release of immunomodulatory agents to enhance T-cell infiltration into tumours post-vaccination and how to sustain the functionality of T cells within the immunosuppressive tumour microenvironment. It is also necessary to examine and validate lymph-node-targeted and APC-targeted delivery of nanovaccines in large animals, as to date most studies have been performed in murine models; this is an important point, as the stability of nanomaterials and lymph-node draining patterns may be entirely different in humans. Also, it is important to streamline and speed up the cGMPs of personalized vaccine nanoparticles for robust adaptability to each patient’s neoantigens, and to establish quality-control measures for neoantigens with diverse physicochemical properties.
Gene-based vaccines
Improvements in technology and in our understanding of diseases has enabled the development of gene therapies as effective and targeted treatments. New approaches for the delivery of DNA and RNA encoding immunostimulatory cytokines and proteins to tumour sites to amplify T-cell responses and reverse immunosuppression within tumours have been developed in the past decades. Here, we discuss the primary methods of gene delivery based on viral and non-viral methods, the latter encompassing a much larger variety of transport mechanisms.
Viral vectors
Gene delivery using viral vectors is effective, owing to the natural ability of viruses to invade host cells and to elicit potent and long-lasting T-cell responses by inducing the durable intracellular expression of antigens. Viral constructs, such as recombinant adenoviruses, vaccinia viruses and fowlpox viruses60,61, have been used to deliver TAAs, including gp100 or MART-1 (melanoma antigen recognized by T cells-1), to patients with metastatic melanoma participating in clinical trials19,62,63,64. Also, in studies of patients with glioblastoma, the intratumoural injection of a herpes simplex virus65 expressing the cytokines IL-12 and IL-4 led to the recruitment of CD4+ and CD8+ T cells as well as macrophages in the tumour site65,66,67,68. A vaccine formulation of a self-replicating Sindbis virus construct linking RNA encoding E7 (a human papilloma virus (HPV) antigen) and VP22 (a herpes symplex virus (HSV) antigen known to facilitate the cellular transport of proteins) also induced a higher frequency of potent IFN-γ-producing splenic CD8+ T cells with enhanced antigen-specific cytotoxicity compared with either E7 or VP22 RNA alone in mice69.
Despite the promise of viral vectors for the delivery of vaccine antigens, identifying the optimal formulations for personalized immunotherapy remains elusive. First, therapies based on viral vectors can result in immune rejection, owing to the induction of immune responses to the viral vectors themselves that reduce their effectiveness. Second, it is technically challenging to produce viral vectors tailored to a patient’s neoantigens that meet cGMP and quality-control standards in a short timeframe, as is required for personalized immunotherapy. In fact, the efficacy of viral vectors for vaccination against neoantigens in humans remains to be seen.
Non-viral delivery
Non-viral gene delivery offers potential solutions to the limitations of viral-vector-based vaccines, as exemplified by the reports of optimized DNA-based gene-delivery systems developed over the past few decades. Direct injection of naked DNA plasmid in mice via the intramuscular, intradermal or intravenous routes enables the transfection of the gene of interest into muscle, skin and liver tissue, respectively70, but the in vivo transfection efficiency of naked DNA is limited by its chemical instability, susceptibility to nuclease attack, rapid clearance and inefficient delivery to local lymph nodes71. Cationic lipids have been widely used to form liposomal complexes with DNA for increased transfection72,73,74, and new delivery systems such as transdermal patches can enhance the targeted delivery of DNA plasmids to skin-resident DCs75,76. Furthermore, clinical trials using DNA-delivery technologies, such as gene guns and electroporation, for vaccine applications are ongoing. Of note, whereas these existing systems should be readily adapted for personalized neoantigen vaccination, DNA immunization generally induces weak immune responses and thus requires co-stimulation with potent adjuvants or with DNA-encoded immunostimulatory genes77,78. Moreover, the potential for induction of anti-DNA antibodies could cause toxicities, and the potential for integration of DNA into the host’s genome is a major concern77,78, especially as neoantigen vaccines would require multiple immunizations with DNA encoding a wide range of antigens.
In the past few years, mRNA-based vaccines have gained much attention owing to a few key advantages over DNA vaccination79,80,81. A major benefit is the lower barrier for antigen expression; when compared with DNA, which requires nuclear delivery, the mRNA-mediated expression of antigens can be achieved via cytosolic delivery. Additionally, mRNA induces only transient gene expression, and is not at risk of genomic integration. Furthermore, RNA sequences can be designed to trigger innate immune responses via interaction with TLRs, retinoic-acid-inducible protein-I (RIG-I), or melanoma-differentiation-associated gene 5 (MDA5)82,83—an inherent immunostimulatory property that makes RNA an attractive technology for vaccine development. However, the chemical instability and low transfection efficacy of mRNA remain major barriers to therapeutic efficacy. Yet, even direct vaccination via intranodal injection of naked mRNA encoding OVA has led to CD8+ and CD4+ T-cell expansion in a B16F10–OVA melanoma model84 and to a first-in-man phase-I clinical trial of mRNA encoding tumour neoantigens identified in patients with stage-III and stage-IV melanoma28. In this clinical study, thirteen patients with late-stage melanoma received ultrasound-guided intranodal vaccinations with up to 20 doses of mRNA encoding 10 different neoantigens (based on each patient’s tumour exome). T-cell responses against 60% of the neoantigens encoded by the mRNA vaccine were detected. Importantly, patients whose tumours expressed known TAAs were vaccinated with both TAAs and neoantigens, but stronger responses were induced against the neoantigens, suggesting that the lack of immune tolerance to the neoantigens is a major factor in the immune response following vaccination. Over 75% of patients remained progression-free for more than a year following neoantigen–mRNA vaccination, and no vaccine-related adverse events were reported. This proof-of-concept clinical study exemplified the promise of mRNA vaccines for personalized cancer immunotherapy28.
Despite this progress, the in vivo delivery of naked mRNA remains challenging. The ribose sugar backbone of RNA, unlike the deoxyribose sugar backbone in DNA, is prone to hydrolysis, which reduces the stability of RNA molecules in circulation. Mammalian mRNAs are on average ~2,000 nucleotides long, and a single event of hydrolysis along the mRNA backbone can impede its translation. Furthermore, ubiquitous ribonucleases within the body decrease the stability of RNA and reduce its therapeutic efficacy. A potential solution for this limitation is the use of synthetic nucleotide analogues or of biomaterial-based delivery for protecting the mRNA from degradation. For example, liposomes encapsulating synthetic mRNA encoding either self-antigens or neoantigens have been efficacious in multiple tumour models, including lymphoma, colon carcinoma and melanoma26,85, as the intravenous administration of mRNA–liposome constructs can lead to their preferential uptake by macrophages and DCs for effective tumour regression and prolonged survival85. Notably, the intravenous administration of mRNA–liposome complexes in a phase-I trial in patients with melanoma induced IFN-α secretion and strong antigen-specific T-cell responses in three of the treated patients85. Also, mRNA-encapsulating cationic liposomes triggered enhanced tumour-specific T-cell expansion and slowed tumour growth in the B16F0 melanoma mouse model as well as in a murine model of glioma86.
Early-stage clinical trials and preclinical evidence have tested the safety and immunogenicity of mRNA vaccines based on neoantigens identified from tumour biopsies. Moreover, the synergy of mRNA neoantigen vaccines and immune-checkpoint blockade offers a clinically suitable pathway for promoting T-cell survival and for augmenting the magnitude and potency of anti-tumour immune responses. Nevertheless, the further refinement of mRNA-vaccine formulations is necessary for their clinical translation. An optimal lipid composition for effective cytosolic delivery of mRNA to human APCs (including DCs, B cells and macrophages) is yet to be identified. Murine studies have screened for lipid compositions that enable the specific targeting of mRNA–liposomes to the spleen rather than the liver85, but whether these findings can be directly translated to humans is unclear. The intravenous injection of mRNA complexes may increase the risk of embolic stroke, lead to off-target deposition of mRNA in the lungs and liver, and promote protein expression in unintended target cells (such as hepatocytes), which could lead to the inadvertent T-cell-mediated killing of host cells. Thus, the composition and chemistry of mRNA-delivery agents should be optimized for balanced mRNA complexation, for selective uptake by the target APCs, and for the unpacking of mRNA within the cytosol for effective antigen expression, with minimal off-target accumulation. In this regard, targeting moieties such as cell-specific peptides may be incorporated into nanoparticles to promote APC-targeted delivery. Also, their co-delivery with adjuvant molecules that synergize with the inherent immunostimulatory functions of RNA should be explored.
Patient-specific cell therapies
The recent advances in gene sequencing and gene editing enable the design of effective patient-specific cell therapies. Attempts to augment cell therapies with the use of biomaterials are particularly promising. Here, we dicuss recent reports on patient-specific cell therapies using DCs and T cells for the treatment of cancer.
DC vaccines
Personalized DC vaccines traditionally involve the differentiation of monocytes isolated from the patient’s peripheral blood, and incubation with tumour antigens or with autologous tumour lysate87,88,89. In one study, among 10 paediatric patients with metastatic solid tumours (including neuroblastoma, osteosarcoma and renal cell cancer) and treated with biweekly DC vaccination, five had stable disease and one patient showed significant tumour regression89; the responders also had an increase in tumour-lysate-reactive T cells in peripheral blood mononuclear cells. In a similar study of patients with glioblastoma multiforme (GBM), those who received three subcutaneous DC vaccines pulsed with autologous GBM tumour lysate had significantly prolonged survival (33 months) compared with patients who received radiation therapy (7.5 months)87; a majority of the vaccinated patients also produced tumour-lysate-specific CD8+ cytotoxic T lymphocyte (CTL) responses, and a subset of responders showed intratumoural CTL infiltration. Vaccination with autologous DCs pulsed with oxidized autologous whole-tumour lysate has also been tested in patients with recurrent ovarian cancer90; here, tumour cells were oxidized before lysis by freeze-thaw cycling, on the basis of previous findings that had shown that this preparation method yielded more effective T-cell priming. DCs pulsed with oxidized tumour lysate were then administered in combination with bevacizumab (a vascular endothelial growth factor A (VEGF-A) antibody inhibitor) and cyclophosphamide to minimize neovascular growth and tumour-infiltration of TREG cells, respectively; of the patients whose T cells responded to the DC vaccine, 73% experienced remission, and 100% reached the 2-year-survival point (only 25% of the non-responders reached it). Immune responses against TAAs as well as previously unidentified neoantigens were observed90. Although lysate-pulsed DC vaccines are safe, well-tolerated and have improved survival outcomes, they have yet to show complete tumour regression or remission91,92. This limitation has spurred efforts towards the engineering of DC vaccines loaded with neoantigens that can elicit tumour-specific T-cell responses with potent immunotherapeutic effects.
A phase-I clinical trial in 2015 that involved three patients with melanoma who had previously received anti-CTLA-4 therapy used a systematic approach for generating DC vaccines loaded with neoantigens93. First, exome sequencing was performed on the patients’ tumours to identify neoepitopes with missense mutations; the hits were then filtered by predictive human leukocyte antigen (HLA)-binding-affinity tools and gene-expression analyses. Neoantigen-pulsed DCs were then administered to the patients. Three out of six neoantigens included in each patient’s regimen led to significant neoantigen-specific CD8+ T-cell expansion. Furthermore, neoantigen–DC vaccination increased the frequency of T-cell receptor (TCR)-β clonotypes that were previously known, and also revealed new TCR-β clonotypes, indicating that the vaccine could activate tumour-specific T cells that otherwise may have been dormant93. This top-down strategy enabled the preparation of DC vaccines tailored to the patients’ tumour mutanome, with maximal therapeutic effect and without toxicity.
Despite these promising results, the successful translation of DC vaccines to patients with reliable outcomes has been challenging, partially because highly trained personnel with specialized equipment are required for the ex vivo differentiation of peripheral blood monocytes into DCs, and for the pulsing of DCs with antigens18,19. In addition, the yield of DCs presenting MHC–peptide complexes is variable and depends on the condition of the patient’s peripheral blood monocytes. Furthermore, once administered, only a minor fraction (<4%) of the injected DCs home to lymphoid tissues94. These limitations, however, could be addressed with new engineering approaches. For example, in situ vaccination with injectable alginate hydrogels, termed ‘cryogels’, can prime local DCs and trigger antitumour immune responses without the need for the ex vivo manipulation of DCs (refs. 95,96,97). Following in vivo implantation, pore-forming cryogels carrying GM-CSF, CpG and tumour antigens recruited immature DCs from local tissues into the scaffold, and promoted the activation of DCs and their migration to draining lymph nodes. In turn, reprogrammed DCs elicited anti-tumour T-cell responses and extended animal survival in murine models of B16F10 melanoma and of breast cancer.
An alternative engineering solution for improving DC vaccines uses exosomes carrying both membrane proteins and endosomal-cell contents. Culturing DCs with exosomes can promote antigen presentation by DCs through the naturally occurring membrane exchange between tumour-antigen-containing exosomes and DCs (ref. 98). To further increase the DC uptake of tumour-cell-derived exosomal vesicles, DCs can be engineered to express extracellular vesicle-internalizing receptors specific for tumour-associated proteins, such as human epidermal growth factor receptor 2 (HER2)99. Moreover, acellular DC-based exosome vaccines have recently gained traction100: compared with DC vaccines, exosomes derived from DCs (Dex) can be readily modified to exhibit favourable characteristics, including enriched MHC–antigen complexes, long-term storage and stability, resistance to immunosuppressive signals, and reactivity with multiple types of immune cells, all in the context of specific ligands, receptors and adjuvants that are naturally present in DCs. Indeed, Dex vaccines have been shown to efficiently promote tumour-antigen-specific T-cell proliferation and subsequent tumour regression in murine tumour models, and have entered phase-I and phase-II clinical trials for several cancers, including non-small-cell lung carcinoma, metastatic melanoma and colorectal cancer101,102,103,104. For a subset of patients from each population, the disease remained stable for up to 1 year, an observation that illustrates the promise of Dex-based immunotherapy100. Given the potential benefits of Dex vaccines, exploring new drug-delivery strategies with Dex or DC-derived vesicles for the cell-targeted and tissue-targeted delivery of immunotherapeutic agents could lead to enhanced efficacy, and might be included in patient-tailored strategies for combination cancer immunotherapy105,106.
Adoptive T-cell therapy
Alongside immune-checkpoint blockade, adoptive T-cell therapy (ACT), which involves the ex vivo purification and manipulation of patient-derived T cells for subsequent injection back into the donor patient, has become a mainstream cancer immunotherapy107,108,109. ACTs follow several strategies. Tumour infiltrating lymphocytes (TILs)—the first type of tumour-associated T cell to be explored as a potential therapy—are isolated from a patient’s tumour biopsies and are expanded ex vivo by using IL-2 stimulation. Although TILs have potential advantages in terms of tumour specificity, they have generally shown limited therapeutic efficacy, prompting the development of other categories of ACT. Two other strategies based on peripheral T cells, transfected with either a transgenic TCR or a chimaeric antigen receptor (CAR), have evolved. By enforcing the expression of an antigen receptor of choice, large batches of cells specific for individual tumour antigens can be produced. Although TCR transgenic T cells and CAR-T cells share similar methodologies, the extracellular single-chain variable fragment (scFv) of CAR-T cells can bind to antigens independently of MHC-mediated presentation on APCs, enabling CAR-T cells to target a wider range of TAAs. As a result of this flexibility, the efficacy of CAR-T cell therapies in treating haematological malignancies has resulted in two FDA-approved therapeutics, tisagenlecleucel and axicabtagene ciloleucel, for the treatment of acute lymphoblastic leukaemia and large B-cell lymphoma, respectively110.
A major factor in the success of T-cell-engineering approaches is the antigen specificity of the inserted antigen receptor. If the target antigen is shared by normal and cancer cells alike, the transferred T cells can cause severe on-target, off-tumour toxicity107; therefore, how to generate CAR-T cells that are as specific to cancer cells as possible to minimize adverse effects and increase efficacy is a major research focus. One approach to enhance cancer-cell specificity involves the development of patient-tailored neoantigen-specific T cells. Indeed, a higher number of putative neoantigens in tumours is known to lead to better prognosis for patients receiving ACT (ref. 111), suggesting that ACT could become more efficacious when T cells are targeted against those neoantigens. This hypothesis is supported by findings from a preclinical study based on T cells transduced with a gene encoding an HLA-specific and mutation-specific TCR for a shared neoantigen that harboured an H3.3K27M mutation (amino acid substitution from lysine to methionine at position 27 of H3.3) from diffuse intrinsic pontine gliomas; infusion of the TCR-engineered T cells led to tumour growth inhibition in a xenograft mouse model112.
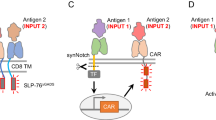
Another limitation of current ACTs is the need to generate a sufficient number of cells ex vivo that can exert a potent therapeutic effect when infused back into the patient. Ex vivo T-cell expansion is a labour-intensive process that requires specialized skills, which limits the availability of ACT to only a few institutions worldwide. A potential solution is the in situ transfection of CAR genes in peripheral T lymphocytes (Fig. 5)113. Circulating T cells can be targeted in vivo by CD3-directed nanoparticles carrying genes for a CD19 CAR to induce its sustained transcription; this strategy led to significantly improved survival of mice bearing Eμ-ALL01 leukaemia cells. Targeting circulating T cells in vivo may also be applicable for generating neoantigen-specific T cells, but the potential for indirect immune activation and the biocompatibility of the proposed system should be carefully assessed.

Plasmid DNA encoding a CAR, a microtubule-associated sequence, a nuclear-localization signal peptide and poly(beta-amino ester) polymer make up the scaffold of the nanoparticle, which is covered with PGA-tailed anti-CD3𝛇 f(ab’)2 via electrostatic interactions. The two plasmids encode an all-murine 194-1BBz CAR and the hyperactive iPB7 transposase. Scale bar, 100 nm. EF1A, eukaryotic translation elongation factor 1 alpha 1; BGH PA, bovine growth hormone polyadenylation signal; AMP, ampicillin resistance gene; ORI, origin of replication. Figure reproduced from ref. 113, Springer Nature Ltd.
An additional strategy to address the current limitations of ACT is to develop artificial APCs (aAPCs) for a more efficient expansion of functional T cells during ex vivo proliferation. Paramagnetic nanoparticles have been modified with agonistic antibodies for providing the activating signals 1 and 2 that enhance T-cell activation and proliferation114. Interestingly, the application of a magnetic field to the nanoparticle–T-cell suspension led to the clustering of the nanoparticles tethered on the cell surface, a process that mimics the signal microclustering observed during the physiological activation of T cells and that enabled the on-demand control of co-stimulatory signals. Similarly, the activation and proliferation of CD8+ T cells has been achieved by means of carbon nanotubes presenting MHC-I molecules and anti-CD28 antibodies, together with poly(lactic-co-glycolic acid) (PLGA) nanoparticles for the delivery of IL-2 (ref. 115). Notably, this system significantly reduced the amount of soluble IL-2 needed to obtain a comparable number of T cells with respect to some commercially available systems for in vitro T-cell expansion. Alternatively, MSRs carrying IL-2, anti-CD3 antibody and peptide-loaded MHC can also promote the expansion of antigen-specific CAR-T cells116. Interestingly, the slow release of IL-2 has emerged as an important factor in T-cell expansion, as it limits the exhaustion of the adoptively transferred T cells, and enhances their function and efficacy.
Maintaining the viability of adoptively transferred T cells and preserving their tumour-infiltration properties are additional challenges for ACT, especially when attempting to improve its efficacy against solid cancers. Owing to the immune-suppressive nature of the tumour microenvironment, it is critical to ensure that the transferred T cells proliferate and home to tumours. To this effect, the surface of CD8+ T cells has been modified with lipid-based nanoparticles carrying IL-15 super-agonist and IL-21 cytokines for potentiating the proliferation of T cells117. Such localized delivery of cytokines enhanced the survival and proliferation of T cells after adoptive transfer into mice bearing cognate-antigen-expressing tumours, and reduced the systemic immunomodulatory effects typically associated with the bolus administration of free cytokines. In a similar approach, lipid-based nanoparticles carrying the topoisomerase I inhibitor SN-38 were attached to the surface of CD8+ T cells118, facilitating their expansion ex vivo while maintaining their lymph-node-homing receptors CD62L (L-selectin) and C–C chemokine receptor type 7 (CCR7). This strategy enabled, in a lymphoma model, the efficient homing of transferred T cells to lymph nodes following administration, the increased delivery of SN-38 by 90-fold as compared with the bolus injection of the drug at 10-fold higher dose and extended animal survival118. Alternatively, adoptively transferred T cells have been targeted in situ with antibody-decorated liposomes carrying IL-2 or a transforming growth factor (TGF)-β inhibitor, which promoted the proliferation of T cells in vivo while reversing the immune-suppressive tumour microenvironment119,120. These biomaterial strategies enable the specific targeting of drugs to the transferred cells, avoiding off-target immunomodulation and thereby safely augmenting the efficacy of T-cell therapies.
Biopolymers can also support T-cell survival and proliferation after adoptive transfer. Alginate-based hydrogels loaded with T cells have been developed for surgical implantation near non-resectable tumours or into the resection cavity following tumour excision121,122. Hydrogels modified with a collagen-mimicking peptide (to promote the efferent migration of T cells) and co-loaded with silica microparticles carrying IL-15 super-agonist and anti-CD3, anti-CD28, and anti-CD137 antibodies (for triggering T-cell proliferation) improved the proliferation of the transferred T cells and enhanced survival in tumour-bearing mice, compared with the systemic administration of the T cells. As biomaterial-based approaches can accommodate and deliver therapeutic cells or immunomodulatory drugs in a localized manner, they minimize T-cell activity in off-target tissues. A few hydrogel-based therapies have been approved by the FDA for bone regeneration, cancer treatment123 and other applications, which is a clear sign of the translational potential of biomaterial-based ACTs.
Emerging combination immunotherapies
Engineering technologies are being investigated to further augment the therapeutic efficacy of immunotherapies. Here, we discuss how harnessing the power of imaging and light modalities can improve patient outcomes using both existing and investigative cancer therapies.
Image-guided theranostics
There are significant obstacles to the targeting of nanoparticles to specific tissues and to the optimization of their tissue permeation, especially for applications in immunomodulation, owing to the potential for organ inflammation and tissue damage associated with drug accumulation in off-target sites. Biomaterial-based image-guided methods, including magnetic resonance imaging (MRI), ultrasound and positron emission tomography–computed tomography (PET–CT), may address these issues by precisely controlling the timing and location of drug release124,125,126,127,128,129, potentially limiting the off-target toxicity observed in combination immunotherapies. In addition, image-guided delivery technologies may provide solutions for enhancing cell permeability and nanoparticle uptake, allowing for better characterization of the tumour microenvironment during and after immunotherapy.
Recent innovations in image-guided theranostics for cancer treatment can be directly adapted to combination immunotherapy, and may lead to new biomaterial-based treatment options for personalized diagnostics and therapeutics. For example, superparamagnetic iron oxide nanoparticles (SPIONs) have been thoroughly investigated as theranostic nanomaterials for MRI applications. SPIONs can be formulated in a variety of sizes and functionalized with different therapeutic moieties by using simple chemistry. They can be used for early tumour diagnosis130,131, for thermal ablation132,133,134 and for the magnetic guidance of therapeutics135,136,137. Importantly, SPIONs are generally biocompatible, as they are readily metabolized to iron ions and oxygen molecules. SPIONs decorated with single-chain anti-CD3 antibodies and carrying immunosuppressive genes have been shown to selectively transfect T cells, leading to the downregulation of cytokine production and proliferation124. And the administration of SPIONs decorated with heat-shock protein (hsp)70 and carrying C6 glioma antigens can target DCs and generate anti-tumour T cells that inhibit tumour growth and extend the survival of C6-glioma-bearing rats138. These two studies of the immunosuppressive or immunostimulant capabilities of SPION-based theranostics, exemplify their potential for individualized combination immunotherapy.
Focussed ultrasound applied to a target site can be used alongside contrast agents that deliver drugs to specific tissues, and thereby potentiate the permeability of the tissues to facilitate the delivery of a drug. Focused ultrasound can also mediate the thermal ablation of tumour tissues. Microbubbles have been used as ultrasound contrast agents (UCAs) for decades in clinical diagnostics, owing to their ability to backscatter sound waves, and have attracted considerable interest as drug-delivery vehicles owing to their ability to produce enough shear force during stable oscillations or acoustic collapse to permeabilize cell membranes139. Microbubbles, similarly to SPIONs, can be functionalized with a variety of therapeutic molecules; for instance, the ultrasound-mediated delivery of chemotherapeutics to different solid tumours has led to tumour regression140,141,142,143. Ultrasound has also been combined with the systemic administration of immunomodulatory agents (to augment their accumulation in specific tissues) in several tumour models, such as a rat glioma model (using soluble IL-12), a mouse hepatoma model (combined with thymidine kinase), and lymphoma, neuroblastoma, melanoma and ovarian-cancer mouse models142,144,145,146.
Integrated PET–CT imaging can predict treatment outcomes, as illustrated by clinical studies showing that tumour-uptake patterns of a glucose analogue are predictive of treatment success147,148,149,150,151. Individual treatment plans can thus be adjusted according to the PET–CT results to improve tumour regression and patient survival. Together with MRI and ultrasound, PET–CT image-guided approaches for the personalization of cancer treatment can be used for the real-time targeting of tumours, and can be combined with other strategies compatible with immunotherapy, including checkpoint blockade and chemotherapy152. The clinical translation of these strategies will be limited mainly by heterogeneity in the patients’ tumours and by difficulties in streamlining treatment procedures for specific tumour locations and tumour burden.
Other combination therapies
A major challenge in cancer immunotherapy is its typically limited efficacy when applied to large tumours. Although in most cases the prioritized therapeutic procedure is the surgical removal of the primary tumour, direct surgical removal is not always feasible. In this context, photothermal therapies (PTTs) may offer an alternative. PTTs use photothermal agents or gold nanoparticles (GNPs) that generate heat once irradiated with near-infrared (NIR) light; when photothermal agents or GNPs are delivered to tumours, NIR-laser irradiation can be locally applied to generate heat in a spatially restricted process, which then ablates the tumour. A similar approach is photodynamic therapy (PDT), which uses photosensitizers to produce reactive oxygen species (such as singlet oxygen,1O2) that can induce apoptosis in cancer cells. Heat or reactive oxygen species can induce ‘immunogenic’ cancer cell death, whereby cancer-specific antigens are released and taken up by APCs, which then trigger cancer-antigen-specific immune responses. Importantly, immune responses triggered by PTT and PDT can synergize with chemotherapies and immunotherapies. For example, the photoablation of tumours using PLGA nanoparticles carrying indocyanine green (ICG), a photothermal agent, and imiquimod, a TLR-7 agonist, in subcutaneous models of primary breast (4T1) and colon (CT26) cancer in mice led to the inhibition of the growth of secondary distant tumours after the direct treatment of the primary tumours with ICG–imiquimod-loaded PLGA nanoparticles, followed by PTT and CTLA-4 checkpoint blockade therapy14. This combination therapy decreased TREG-cell numbers while increasing T-cell infiltration in secondary tumours, resulting in the regression of primary tumours and the suppression of secondary tumours. Also, PTT combined with chemotherapy can trigger potent systemic anti-tumour immunity against disseminated, untreated tumours (Fig. 6)17. Specifically, the treatment of a primary tumour with a single round of PTT with polydopamine-coated spiky gold nanoparticles153 and a sub-therapeutic dose of doxorubicin elicited robust anti-tumour responses in CD8+ T-cells and natural-killer (NK) cells, eliminating local as well as untreated distant tumours in over 85% of mice bearing CT26 colon carcinomas. The approach was also efficacious in a highly aggressive model of advanced head-and-neck squamous cell carcinoma (HNSCC), a murine model that closely mimics the clinical trials of PTT with silica–gold nanoshells17.

A polydopamine (PDA) coating on spiky gold nanoparticles (SGNP–PDA) prevents their thermal deformation, improving photothermal efficiency (left). The intratumoural injection of SGNP–PDA followed by laser irradiation induces the release of tumour antigens and danger signals that lead to DC maturation while promoting the secretion of stress-inducible ligands by the cancer cells (middle). Together, these activate and stimulate the proliferation of CD8+ T and NK cells, leading to systemic immune responses (right). Figure reproduced from ref. 17, Springer Nature Ltd.
PDT can also trigger systemic immune responses. For instance, core–shell structured coordination-polymer nanoparticles loaded with oxaliplatin (a chemotherapeutic) and with photosensitizer pyrolipid surrounding the core enabled dual chemotherapy and PDT, inhibiting tumour growth in murine models of syngeneic CT26 and xenograft HT29 cancer-cell lines154. Anti-PD-L1 treatment further potentiated anticancer efficacy in bilateral MC38 and CT26 mouse tumour models; intraperitoneal injections of the nanoparticles in tumour-bearing mice, followed by light irradiation only at the primary tumour and by the systemic administration of anti-PDL-1 antibody, reduced the growth of both primary and secondary tumours, with evidence of an abscopal effect. In a similar context, radiotherapy and radiodynamic therapy were applied via a Hafnium (Hf) nanoscale metal-organic framework (nMOF), an X-ray scintillator that can be excited with X-rays to generate ionizing radiation for tumour killing (Fig. 7)155. This study used 5,15-di(p-benzoato)porphyrin (DBP) as a photosensitizer ligand to crosslink Hf-based nMOF clusters, which generated OH radicals following X-ray radiation and excited the photosensitizer with ionizing radiation to generate singlet oxygen. In addition, DBP-crosslinked Hf-nMOF (DBP–Hf) was loaded with an indoleamine 2,3-dioxygenase inhibitor (IDOi), INCB024360 (also known as epacadostat), to exert immunotherapeutic effects. Intratumoral injection of DBP–Hf or systemic injection of PEGylated DBP–Hf, followed by X-ray radiation at the tumour site, led to rapid tumour regression in various human-cancer xenograft models and in a murine CT26 colorectal-cancer model. Together with an IDOi, IDO/DBP–Hf eradicated primary tumours and inhibited distant tumour growth in subcutaneous bilateral cancer models of CT26 and TUBO breast cancer cells155. Another approach involved antigen-capturing nanoparticles and radiotherapy for the capture of melanoma neoantigens released by irradiated tumour cells, leading to the expansion of CD8+ T cells, to increases in the CD4+/TREG and CD8+/TREG ratios, and to the regression of B16F10 melanoma tumours156.

Intratumoural injection, into the primary tumour, of the immune-checkpoint agent IDOi encapsulated in an nMOF, followed by radiation, generates reactive oxygen species that induce tumour-cell death and antigen release. In turn, the antigen is presented to T cells by DCs, leading to T-cell activation and proliferation. The potent systemic immune response results in the eradication of a distal secondary tumour. Figure reproduced from ref. 155, Springer Nature Ltd.
Collectively, these studies support the use of PTT, PDT and radiotherapy for the eradication of primary tumours and for triggering the release of tumour antigens (including neoantigens), endogenous danger signals and pro-inflammatory cytokines within the tumour microenvironment. Such changes in the tumour microenvironment may provide an individualized treatment regimen for treating primary tumours and also for initiating immune responses, especially in combination with checkpoint-blockade therapy and with other immunostimulatory agents. In fact, recent studies have examined the clinical efficacy of radiation combined with the checkpoint inhibitor ipilimumab157,158. Patients with chemoresistant prostate cancer, melanoma or lung cancer benefited from the combination therapy and, notably, a patient with non-small-cell lung carcinoma who had no response to prior chemotherapies experienced a complete response to the combination therapy. These findings suggest that such combination therapy can turn non-immunogenic tumours into immunologically active ones, and that it may be feasible to further bolster their anti-tumour effects via theranostic approaches that enable spatiotemporal control over the delivery of adjuvants.
Still, synergies between cancer immunotherapy and PTT, PDT or radiotherapy are far from optimal. For example, retrospective analyses of clinical studies of such combination therapies strongly suggest the need to optimize radiation conditions to elicit strong T-cell priming in the presence of immune-checkpoint inhibitors159,160. Photosensitive and radiosensitive materials, especially metal-based agents, will need to be thoroughly evaluated for biocompatibility. Most agents for PTT and PDT need to be delivered to tumours for effective therapy; therefore, approaches to increase their tumour accumulation remain a major research focus. Chemically modifying photosensitive and radiosensitive materials or encapsulating them into biomaterials may reduce their toxicity and increase their in vivo biocompatibility and stability. Naturally, the clinical application of these techniques will require thorough dose–response studies, to determine tolerated levels of skin irritation as well as any other local and systemic adverse events. Moreover, it remains unclear how PTT, PDT and radiotherapy differ in terms of the mechanisms of action underpinning their ability to elicit anti-tumour immunity, and what their impact on immunosuppression is. Hence, systematic studies are warranted to delineate the local and systemic effects of each modality in order to fully exploit their impact on immune activation.
Translational challenges and opportunities
The studies and engineering approaches that we have here highlighted illustrate the potential of personalized immunotherapy. Yet, many clinically relevant technical and scientific considerations remain to be addressed.
Inter-patient heterogeneity
Mutational loads differ between cancer types as well as between patient populations20,161. This calls for bioinformatics tools that can predict top neoantigen candidates for specific cancer types20,162,163. Also, the physicochemical properties of neoantigens need to be considered as criteria in the nomination of neoantigen candidates, as they directly impact the delivery of neoantigens to APCs in draining lymph nodes in vivo, and thereby affect the overall immunogenicity and efficacy of personalized vaccines. Additionally, one needs to also consider the competitive binding of candidate neoantigens to MHC molecules, as well as combinations with other antigens in multi-antigen delivery systems, in order to achieve broad T-cell responses. Furthermore, engineering approaches that enable the expeditious screening of the reactivity of patient specimens against neoantigens should be explored; for example, the use of a blood biopsy for the high-throughput screening of top neoantigen candidates164, or for whole-exome sequencing of the patient’s cell-free DNA (ref. 165), may facilitate neoantigen discovery and identification.
Personalized-therapy testing
After nomination of neoantigens, target validation may also benefit from new engineering tools. Recent advances in organ-on-chip technologies166 can be applied to build patient-derived tumours for testing and validating the specificity and functionality of neoantigen-specific T cells (for example, at the level of tumour infiltration and killing). Furthermore, as immune-checkpoint blockade frequently causes immune-related adverse events in gut, skin, endocrine glands, liver and lung167, patient-tailored immunotherapies may be tested for their safety profiles using organ-on-a-chip technologies tailored to each organ.
Manufacturing
Any strategy for improving personalized immunotherapy must be easily adoptable, with streamlined manufacturing procedures. This is a crucial factor for clinical translation, in particular because a current bottleneck in personalized cancer immunotherapy is the extensive time required for the cGMP production of personalized vaccine products. For instance, in the first-in-human application of neoantigen-encoding mRNA vaccines, the time required for neoantigen selection from patients, mRNA production, vaccine formulation and product release, ranged from 89 to 160 days, with a median time of 103 days28. Similarly, the development of personalized neoantigen peptide vaccines took a median time of 18 weeks from tumour biopsy to vaccine administration27. The demands of patients with advanced cancer should thus be met with simplified materials design and robust formulation protocols, avoiding convoluted systems that add unnecessary layers of complexity or that pose significant challenges for patient-specific manufacturing and product release. In this regard, developments in the microfluidic-based fabrication of microparticles and nanoparticles could lead to standardized manufacturing solutions168,169,170. Microfluidic technologies may enable the production of vaccine-delivery vehicles in a well-controlled and high-throughput manner, and their smaller scale of synthesis may also be suitable for the personalized manufacturing of therapeutics tailored to each patient.
Quality control and safety
Owing to the broad ranges of stability and physicochemical properties of peptides, proteins, biomaterials and plasmids, personalized immunotherapies will require customized formulations that use standardized analytical methods, standardized sterilization procedures and standardized criteria for product release. Moreover, safety and dosage profiles must be established for each therapeutic modality and biomaterial. As different therapeutic systems exhibit different transport kinetics in vivo, lower doses may be needed for therapeutic moieties with faster kinetics or bigger potency in order to achieve the desired therapeutic effect.
Cost
A financial assessment for each treatment modality and combination must accurately account for the cost of treatments, and must consider infrastructure needs and costs associated with individualized immunotherapy, including the cost of the biopsy of the tumour, DNA and RNA sequencing, the use of a bioinformatics pipeline, image-guided diagnostics, the cGMP-manufacturing of patient-specific biologics, cells and drug delivery carriers, and the need of highly skilled medical personnel for patient-tailored dosing and procedures. Because these personalized processes are necessary to maintain the integrity of each immunotherapy, it is imperative that the processes are streamlined and the costs reduced so as to accelerate patient access to these therapies.
Prevention
No less important than designing treatment strategies is to focus these same principles and technical advances on preventive vaccines against pre-malignant conditions171. For instance, patients with Lynch syndrome who have an unusually high rate of mutations would be prime candidates for prophylactic vaccination against tumour development172,173.
Animal models
Most preclinical studies in cancer therapy, including immunotherapy, are performed in rodents; yet, validation in large animal studies is necessary. The general consensus is that murine tumour models do not faithfully recapitulate human-tumour development; and, importantly, clinical failures in cancer nanomedicine are thought to result in part from the heavy dependence on murine models for the early identification and validation of therapies. The design and testing of engineering approaches for immunotherapies should take a page from the clinical failures of earlier efforts in the development of nanomedicines174.
Outlook
Deciding which of the different strategies for personalized cancer immunotherapy are the most appropriate for individual patients will largely depend on the patient’s tumour burden and their specific treatment needs (Table 1). Individually, each immunotherapy has particular advantages: delivering neoantigen peptides via nanoparticles offers a truly patient-specific treatment that is tailored to individual tumours; gene therapy by using nanoparticles or polyplexes to deliver neoantigen-encoding nucleic RNAs and immunostimulatory cytokines has shown promising efficacy results; personalized cellular therapies, including adoptive transfer of genetically engineered T cells, can effectively boost anti-tumour immunity, with effector cells bearing enhanced functions; DC vaccines present neoantigens in vivo to the T cells of an individual patient; and image-guided, theranostic approaches based on biomaterials responsive to magnetic resonance and ultrasound are arising as an adjunct therapy in preclinical studies. Perhaps more importantly, these strategies may be rationally combined to promote synergistic anti-tumour effects and to achieve unprecedented outcomes174. Although immune-checkpoint blockade, ACT and neoantigen vaccines have been individually successful in the clinic, combining these therapies might further improve patient outcomes.
The engineering strategies discussed in this Perspective, including the design of synthetic materials on the basis of desired physicochemical criteria and the targeted delivery of therapeutics via specific biomolecules or via imaging, may enhance each modality’s therapeutic efficacy while allowing for synergies to occur. The clinical success of these therapeutic approaches will depend on their manufacturing feasibility, on consistent outcomes from patient to patient, and ultimately on patient-survival benefits. As the breadth and success of cancer immunotherapies continues to expand, personalized immunotherapies for the management of patients with cancer will not only need to eradicate tumours but also maintain anti-tumour immunity for the remainder of the patient’s life.
References
Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Garon, E. B. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).
Rosenberg, J. E. et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Wolchok, J. D. et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 369, 122–133 (2013).
Demaria, S. et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 11, 728–734 (2005).
Vanneman, M. & Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 12, 237–251 (2012).
Cho, H. I., Barrios, K., Lee, Y. R., Linowski, A. K. & Celis, E. BiVax: a peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol. Immunother. 62, 787–799 (2013).
Duraiswamy, J., Kaluza, K. M., Freeman, G. J. & Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603 (2013).
Belcaid, Z. et al. Focal radiation therapy combined with 4–1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS ONE 9, e101764 (2014).
Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015).
Chen, Q. et al. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 7, 1–13 (2016).
Bethune, M. & Joglekar, A. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr. Op. Biotechnol. 48, 142–152 (2017).
Kuai, R., Ochyl, L. J., Bahjat, K. S., Schwendeman, A. & Moon, J. J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mat. 16, 489–496 (2017).
Nam, J. et al. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 9, 1–13 (2018).
Kugler, A. et al. Regression of human metastatic renal cell carcinoma after vaccination with tumor cell–dendritic cell hybrids. Nat. Med. 6, 332–336 (2000).
Panelli, M. C. et al. Phase 1 study in patients with metastatic melanoma of immunization with dendritic cells presenting epitopes derived from the melanoma-associated antigens MART-1 and gp100. J. Immunother. 23, 487–498 (2000).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Hu, Z., Ott, P. A. & Wu, C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018).
Sahin, U. & Tureci, O. Personalized vaccines for cancer immunotherapy. Science 359, 1355–1360 (2018).
Castle, J. C. et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 72, 1081–1091 (2012).
Yadav, M. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014).
Gubin, M. M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014).
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Hilf, N. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245 (2019).
Bijker, M. S. et al. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J. Immunol. 179, 5033–5040 (2007).
Kakimi, K. et al. A phase I study of vaccination with NY‐ESO‐1f peptide mixed with Picibanil OK‐432 and Montanide ISA‐51 in patients with cancers expressing the NY‐ESO‐1 antigen. Int. J. Cancer 129, 2836–2846 (2011).
van Hall, T. & van der Burg, S. H. in Advances in Immunology Vol. 114 (ed. Melief, C.) 51–76 (Elsevier, 2012).
Hailemichael, Y. et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 19, 465–472 (2013).
Di Stasi, A., Jimenez, A. M., Minagawa, K., Al-Obaidi, M. & Rezvani, K. Review of the results of WT1 peptide vaccination strategies for myelodysplastic syndromes and acute myeloid leukemia from nine different studies. Front. Immunol. 6, 1–6 (2015).
Blander, J. M. & Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440, 808–812 (2006).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010).
Fan, Y. C. & Moon, J. J. Nanoparticle drug delivery systems designed to improve cancer vaccines and immunotherapy. Vaccines 3, 662–685 (2015).
Reddy, S. T. et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 25, 1159–1164 (2007).
Irvine, D. J., Hanson, M. C., Rakhra, K. & Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 115, 11109–11146 (2015).
Shi, J., Kantoff, P. W., Wooster, R. & Farokhzad, O. C. Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017).
Kuai, R., Li, D., Chen, Y. E., Moon, J. J. & Schwendeman, A. High-density lipoproteins: nature’s multifunctional nanoparticles. ACS Nano 10, 3015–3041 (2016).
Cruz, P. M., Mo, H., McConathy, W. J., Sabnis, N. & Lacko, A. G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 4, 1–7 (2013).
Zheng, Y. et al. Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics 3, 477–486 (2013).
Shahzad, M. M. et al. Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles. Neoplasia 13, 309–319 (2011).
Qian, Y. et al. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials 98, 171–183 (2016).
Kuai, R. et al. Subcutaneous nanodisc vaccination with neoantigens for combination cancer immunotherapy. Bioconj. Chem. 29, 771–775 (2018).
Kuai, R. et al. Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J Control Release 282, 131–139 (2018).
Irvine, D. Material aid for vaccines. Nat. Mat. 17, 472–473 (2018).
Tyagi, P. & Santos, J. L. Macromolecule nanotherapeutics: approaches and challenges. Drug Discov. Today 23, 1053–1061 (2018).
Liu, H. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014).
Zhu, G. et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat. Commun. 8, 1–15 (2017).
Dawidczyk, C. M. et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Control Release 187, 133–144 (2014).
Wittrock, S., Becker, T. & Kunz, H. Synthetic vaccines of tumor-associated glycopeptide antigens by immune-compatible thioether linkage to bovine serum albumin. Angew. Chem. Int. 46, 5226–5230 (2007).
Wang, P. et al. An albumin-binding polypeptide both targets cytotoxic T lymphocyte vaccines to lymph nodes and boosts vaccine presentation by dendritic cells. Theranostics 8, 223–236 (2018).
Zhu, G. et al. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat. Commun. 8, 1–13 (2017).
Luo, M. et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 12, 648–654 (2017).
Li, A. W. et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat. Mat. 17, 528–534 (2018).
Wegmann, F. et al. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat. Biotechnol. 30, 883–888 (2012).
Rosenberg, S. A., Yang, J. C. & Restifo, N. P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915 (2004).
Kent, S. J. et al. Enhanced T-cell immunogenicity and protective efficacy of a human immunodeficiency virus type 1 vaccine regimen consisting of consecutive priming with DNA and boosting with recombinant fowlpox virus. J. Virol. 72, 10180–10188 (1998).
Rosenberg, S. A. et al. Recombinant fowlpox viruses encoding the anchor-modified gp100 melanoma antigen can generate antitumor immune responses in patients with metastatic melanoma. Clin. Cancer Res. 9, 2973–2980 (2003).
Kaufman, H. L. et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J. Clin. Invest. 115, 1903–1912 (2005).
Rosenberg, S. A. et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J. Natl Cancer Inst. 90, 1870–1872 (1998).
Parker, J. N. et al. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl Acad. Sci. USA 97, 2208–2213 (2000).
Markert, J. M. et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 17, 199–207 (2008).
Varghese, S. et al. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 13, 253–265 (2005).
Joshi, B. H. et al. In situ expression of interleukin-4 (IL-4) receptors in human brain tumors and cytotoxicity of a recombinant IL-4 cytotoxin in primary glioblastoma cell cultures. Cancer Res. 61, 8058–8061 (2001).
Cheng, W.-F. et al. Enhancement of Sindbis virus self-replicating RNA vaccine potency by linkage of herpes simplex virus type 1 VP22 protein to antigen. J. Virol. 75, 2368–2376 (2001).
Herweijer, H. & Wolff, J. Progress and prospects: naked DNA gene transfer and therapy. Gene Ther. 10, 453–458 (2003).
Kawabata, K., Takakura, Y. & Hashida, M. The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm. Res. 12, 825–830 (1995).
Nomura, T. et al. Intratumoral pharmacokinetics and in vivo gene expression of naked plasmid DNA and its cationic liposome complexes after direct gene transfer. Can. Res. 57, 2681–2686 (1997).
Li, S. & Huang, L. In vivo gene transfer via intravenous administration of cationic lipid-protamine-DNA (LPD) complexes. Gene Ther. 4, 891–900 (1997).
Xu, Y. & Szoka, F. C. Jr. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 35, 5616–5623 (1996).
Cheng, J. Y. et al. Transcutaneous immunization by lipoplex-patch based DNA vaccines is effective vaccination against Japanese encephalitis virus infection. J. Control Rel. 135, 242–249 (2009).
Gill, H. S., Soderholm, J., Prausnitz, M. R. & Sallberg, M. Cutaneous vaccination using microneedles coated with hepatitis C DNA vaccine. Gene Ther. 17, 811–814 (2010).
Rice, J., Ottensmeier, C. H. & Stevenson, F. K. DNA vaccines: precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 8, 108–120 (2008).
Kutzler, M. A. & Weiner, D. B. DNA vaccines: ready for prime time? Nat. Rev. Genet. 9, 776–788 (2008).
Schlake, T., Thess, A., Fotin-Mleczek, M. & Kallen, K. J. Developing mRNA-vaccine technologies. RNA Biol. 9, 1319–1330 (2012).
Geall, A. J., Mandl, C. W. & Ulmer, J. B. RNA: the new revolution in nucleic acid vaccines. Semin. Immunol. 25, 152–159 (2013).
Deering, R. P., Kommareddy, S., Ulmer, J. B., Brito, L. A. & Geall, A. J. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin. Drug Deliv. 11, 885–899 (2014).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Thompson, A. J. & Locarnini, S. A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 85, 435–445 (2007).
Kreiter, S. et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 70, 9031–9040 (2010).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Sayour, E. J. et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. Oncoimmunology 6, e1256527 (2017).
Yu, J. S. et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 64, 4973–4979 (2004).
Kim, C.-H. et al. Enhancement of anti-tumor immunity specific to murine glioma by vaccination with tumor cell lysate-pulsed dendritic cells engineered to produce interleukin-12. Cancer Immunol. Immunother. 55, 1309–1319 (2006).
Geiger, J. D. et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 61, 8513–8519 (2001).
Tanyi, J. L. et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 10, eaao5931 (2018).
Fadul, C. E. et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 34, 382–389 (2011).
Schwaab, T. et al. Clinical and immunologic effects of intranodal autologous tumor lysate-dendritic cell vaccine with aldesleukin (interleukin 2) and IFN-α2a therapy in metastatic renal cell carcinoma patients. Clin. Cancer Res. 15, 4986–4992 (2009).
Carreno, B. M. et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Srinivas, M. et al. Imaging of cellular therapies. Adv. Drug Deliv. Rev. 62, 1080–1093 (2010).
Bencherif, S. A. et al. Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun. 6, 7556 (2015).
Verbeke, C. S. & Mooney, D. J. Injectable, Pore-Forming Hydrogels for In Vivo Enrichment of Immature Dendritic Cells. Adv. Healthcare Mat. 4, 2677–2687 (2015).
Shih, T. Y. et al. Injectable, tough alginate cryogels as cancer vaccines. Adv. Healthcare Mat. 7, e1701469 (2018).
Théry, C., Zitvogel, L. & Amigorena, S. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2, 569–579 (2002).
Squadrito, M. L., Cianciaruso, C., Hansen, S. K. & De Palma, M. EVIR: chimeric receptors that enhance dendritic cell cross-dressing with tumor antigens. Nat. Methods 15, 183–186 (2018).
Pitt, J. M. et al. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Invest. 126, 1224–1232 (2016).
Escudier, B. et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J. Transl. Med. 3, 1–13 (2005).
Morse, M. A. et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 3, 1–8 (2005).
Dai, S. et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 16, 782–790 (2008).
Besse, B. et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 5, e1071008 (2016).
van Dommelen, S. M. et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J. Control Rel. 161, 635–644 (2012).
Johnsen, K. B. et al. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta 1846, 75–87 (2014).
Restifo, N. P., Dudley, M. E. & Rosenberg, S. A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 12, 269–281 (2012).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Tang, J., Shalabi, A. & Hubbard-Lucey, V. M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 29, 84–91 (2018).
Lauss, M. et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 8, 1–11 (2017).
Chheda, Z. et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 215, 141–157 (2018).
Smith, T. T. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 12, 813–820 (2017).
Kosmides, A. K., Necochea, K., Hickey, J. W. & Schneck, J. P. Separating T cell targeting components onto magnetically clustered nanoparticles boosts activation. Nano Lett. 18, 1916–1924 (2018).
Fadel, T. R. et al. A carbon nanotube-polymer composite for T-cell therapy. Nat. Nanotechnol. 9, 639–647 (2014).
Cheung, A. S., Zhang, D. K. Y., Koshy, S. T. & Mooney, D. J. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol. 36, 160–169 (2018).
Stephan, M., Moon, J., Um, S., Bershteyn, A. & Irvine, D. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 16, 1035–1041 (2010).
Huang, B. et al. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 7, 1–21 (2015).
Zheng, Y. et al. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J. Cont. Rel. 172, 426–435 (2013).
Zheng, Y., Tang, L., Mabardi, L., Kumari, S. & Irvine, D. Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or noninternalizing receptors. ACS Nano 11, 3089–3100 (2017).
Smith, T. et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest. 127, 2176–2191 (2017).
Stephan, S. et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 33, 97–101 (2015).
Li, J. & Mooney, D. J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 1, 16071 (2016).
Chen, G. et al. MRI-visible polymeric vector bearing CD3 single chain antibody for gene delivery to T cells for immunosuppression. Biomaterials 30, 1962–1970 (2009).
Fan, Z., Chen, D. & Deng, C. Improving ultrasound gene transfection efficiency by controlling ultrasound excitation of microbubbles. J. Contr. Rel. 170, 401–413 (2013).
Weber-Adrian, D. et al. Gene delivery to the spinal cord using MRI-guided focused ultrasound. Gene Ther. 22, 568–577 (2015).
Chertok, B., Langer, R. & Anderson, D. G. Spatial control of gene expression by nanocarriers using heparin masking and ultrasound-targeted microbubble destruction. ACS Nano 10, 7267–7278 (2016).
Mead, B. P. et al. Targeted gene transfer to the brain via the delivery of brain-penetrating DNA nanoparticles with focused ultrasound. J. Contr. Rel. 223, 109–117 (2016).
Fan, C.-H. et al. Folate-conjugated gene-carrying microbubbles with focused ultrasound for concurrent blood-brain barrier opening and local gene delivery. Biomaterials 106, 46–57 (2016).
Ling, D. et al. Multifunctional tumor pH-sensitive self-assembled nanoparticles for bimodal imaging and treatment of resistant heterogeneous tumors. J. Am. Chem. Soc. 136, 5647–5655 (2014).
Yang, K. et al. Multimodal imaging guided photothermal therapy using functionalized graphene nanosheets anchored with magnetic nanoparticles. Adv. Mat. 24, 1868–1872 (2012).
Lee, J.-H. et al. Exchange-coupled magnetic nanoparticles for efficient heat induction. Nat. Nanotechnol. 6, 418–422 (2011).
Riedinger, A. et al. Subnanometer local temperature probing and remotely controlled drug release based on azo-functionalized iron oxide nanoparticles. Nano Lett. 13, 2399–2406 (2013).
Hayashi, K. et al. Superparamagnetic nanoparticle clusters for cancer theranostics combining magnetic resonance imaging and hyperthermia treatment. Theranostics 3, 366–376 (2013).
Dames, P. et al. Targeted delivery of magnetic aerosol droplets to the lung. Nat. Nanotechnol. 2, 495–499 (2007).
Amirfazli, A. Nanomedicine: magnetic nanoparticles hit the target. Nat. Nanotechnol. 2, 467–468 (2007).
Galanzha, E. I. et al. In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells. Nat. Nanotechnol. 4, 855–860 (2009).
Shevtsov, M. A. et al. 70-kDa heat shock protein coated magnetic nanocarriers as a nanovaccine for induction of anti-tumor immune response in experimental glioma. J. Cont. Rel. 220, 329–340 (2015).
Sirsi, S. & Borden, M. Microbubble Compositions, Properties and Biomedical Applications. Bubble Sci. Eng. Technol. 1, 3–17 (2009).
Treat, L. H. et al. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI‐guided focused ultrasound. Int. J. Cancer 121, 901–907 (2007).
Burke, C. W., Alexander, E., Timbie, K., Kilbanov, A. L. & Price, R. J. Ultrasound-activated agents comprised of 5FU-bearing nanoparticles bonded to microbubbles inhibit solid tumor growth and improve survival. Mol. Ther. 22, 321–328 (2014).
Chen, P.-Y. et al. Focused ultrasound-induced blood–brain barrier opening to enhance interleukin-12 delivery for brain tumor immunotherapy: a preclinical feasibility study. J. Transl Med. 13, 1–12 (2015).
Timbie, K. F. et al. MR image-guided delivery of cisplatin-loaded brain-penetrating nanoparticles to invasive glioma with focused ultrasound. J. Cont. Rel. 263, 120–131 (2017).
Suzuki, R. et al. A novel strategy utilizing ultrasound for antigen delivery in dendritic cell-based cancer immunotherapy. J. Cont. Rel. 133, 198–205 (2009).
Zhou, S. et al. Ultrasound-targeted microbubble destruction mediated herpes simplex virus-thymidine kinase gene treats hepatoma in mice. J. Exp. Clin. Cancer Res. 29, 1–6 (2010).
Unga, J. & Hashida, M. Ultrasound induced cancer immunotherapy. Adv. Drug Del. Rev. 72, 144–153 (2014).
Yap, J. T., Carney, J. P., Hall, N. C. & Townsend, D. W. Image‐guided cancer therapy using PET/CT. Cancer J. 10, 221–233 (2004).
Bundschuh, R. A. et al. Textural parameters of tumor heterogeneity in 18F-FDG PET/CT for therapy response assessment and prognosis in patients with locally advanced rectal cancer. J. Nucl. Med. 55, 891–897 (2014).
Hillner, B. E. et al. 18F-fluoride PET used for treatment monitoring of systemic cancer therapy: results from the National Oncologic PET Registry. J. Nucl. Med. 56, 222–228 (2015).
Connolly, R. M. et al. TBCRC 008: early change in 18F-FDG uptake on PET predicts response to preoperative systemic therapy in human epidermal growth factor receptor 2–negative primary operable breast cancer. J. Nucl. Med. 56, 31–37 (2015).
Norregaard, K. et al. 18F-FDG PET/CT-based early treatment response evaluation of nanoparticle-assisted photothermal cancer therapy. PLoS ONE 12, e0177997 (2017).
Kuai, R. et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 4, eaao1736 (2018).
Nam, J., Son, S. & Moon, J. J. Adjuvant-loaded spiky gold nanoparticles for activation of innate immune cells. Cell. Mol. Bioeng. 10, 341–355 (2017).
He, C. et al. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 7, 1–12 (2016).
Lu, K. et al. Low-dose X-ray radiotherapy-radiodynamic therapy via nanoscale metal-organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng. 2, 600–610 (2018).
Min, Y. et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 12, 877–882 (2017).
Demaria, S., Golden, E. B. & Formenti, S. C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 1, 1325–1332 (2015).
Demaria, S., Coleman, C. N. & Formenti, S. C. Radiotherapy: changing the game in immunotherapy. Trends Cancer 2, 286–294 (2016).
Reynders, K., Illidge, T., Siva, S., Chang, J. Y. & De Ruysscher, D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 41, 503–510 (2015).
Barker, C. A. & Postow, M. A. Combinations of radiation therapy and immunotherapy for melanoma: a review of clinical outcomes. Int. J. Radiat. Oncol. Biol. Phys. 88, 986–997 (2014).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Gubin, M. M., Artyomov, M. N., Mardis, E. R. & Schreiber, R. D. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J. Clin. Invest. 125, 3413–3421 (2015).
Snyder, A. & Chan, T. A. Immunogenic peptide discovery in cancer genomes. Curr. Opin. Genet. Dev. 30, 7–16 (2015).
Gros, A. et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 22, 433–438 (2016).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1–13 (2017).
Bhatia, S. N. & Ingber, D. E. Microfluidic organs-on-chips. Nat. Biotechnol. 32, 760–772 (2014).
Michot, J. M. et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54, 139–148 (2016).
Shestopalov, I., Tice, J. D. & Ismagilov, R. F. Multi-step synthesis of nanoparticles performed on millisecond time scale in a microfluidic droplet-based system. Lab Chip 4, 316–321 (2004).
Jahn, A., Vreeland, W. N., DeVoe, D. L., Locascio, L. E. & Gaitan, M. Microfluidic directed formation of liposomes of controlled size. Langmuir 23, 6289–6293 (2007).
Valencia, P. M., Farokhzad, O. C., Karnik, R. & Langer, R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat. Nanotechnol. 7, 623–629 (2012).
Finn, O. J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 18, 183–194 (2018).
Bonadona, V. et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305, 2304–2310 (2011).
von Knebel Doeberitz, M. & Kloor, M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam. Cancer 12, 307–312 (2013).
Nam, J. et al. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mat. 4, 398–414 (2019).
Acknowledgements
This work was supported in part by NIH (grant no. R01EB022563, R01CA210273, R01CA223804, R01AI127070, R21NS091555, R01HL134569, U01CA210152, R37NS094804, RO1NS105556, R21NS107894, RO1NS096756, UO1CA224160), MTRAC for Life Sciences Hub, UM Forbes Institute for Cancer Discovery Pilot Grant and Emerald Foundation. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (grant no. 348774), DoD/CDMRP Peer Reviewed Cancer Research Program (grant no. W81XWH-16-1-0369), and NSF CAREER Award (grant no. 1553831). L.S. acknowledges financial support from the UM Pharmacological Sciences Training Program (PSTP) (grant no. GM007767 from NIGMS). K.S.P. acknowledges financial support from the UM TEAM Training Program (grant no. DE007057 from NIDCR). Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defence.
Author information
Authors and Affiliations
Contributions
L.S., K.S.P, and J.J.M. discussed content, researched data and wrote the manuscript. Q.L., P.R.L, M.G.C. and A.S. contributed to the discussion.
Corresponding author
Ethics declarations
Competing interests
A patent application for the nanodisc vaccines has been filed, with J.J.M. and A.S. as inventors, and J.J.M. and A.S. as co-founders of EVOQ Therapeutics, LLC., which develops the technology.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scheetz, L., Park, K.S., Li, Q. et al. Engineering patient-specific cancer immunotherapies. Nat Biomed Eng 3, 768–782 (2019). https://doi.org/10.1038/s41551-019-0436-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-019-0436-x
This article is cited by
-
Nucleic acid-based drugs for patients with solid tumours
Nature Reviews Clinical Oncology (2024)
-
Harnessing 3D in vitro systems to model immune responses to solid tumours: a step towards improving and creating personalized immunotherapies
Nature Reviews Immunology (2024)
-
Nanoparticle delivery for central nervous system diseases and its clinical application
Nano Research (2024)
-
Neoantigens: promising targets for cancer therapy
Signal Transduction and Targeted Therapy (2023)
-
Overcoming tumor and mucosal barriers through active-loaded nanocarriers: nanoparticles and exosomes
Applied Nanoscience (2023)
