
Abstract
A malaria vaccine that prevents infection will be an important new tool in continued efforts of malaria elimination, and such vaccines are under intense development for the major human malaria parasite Plasmodium falciparum (Pf). Antibodies elicited by vaccines can block the initial phases of parasite infection when sporozoites are deposited into the skin by mosquito bite and then target the liver for further development. However, there are currently no standardized in vivo preclinical models that can measure the inhibitory activity of antibody specificities against Pf sporozoite infection via mosquito bite. Here, we use human liver-chimeric mice as a challenge model to assess prevention of natural Pf sporozoite infection by antibodies. We demonstrate that these mice are consistently infected with Pf by mosquito bite and that this challenge can be combined with passive transfer of either monoclonal antibodies or polyclonal human IgG from immune serum to measure antibody-mediated blocking of parasite infection using bioluminescent imaging. This methodology is useful to down-select functional antibodies and to investigate mechanisms or immune correlates of protection in clinical trials, thereby informing rational vaccine optimization.
Similar content being viewed by others


Introduction
Despite considerable effort and substantial progress in reducing the malaria burden in many countries over the past decade, more than 200 million people still suffered from this parasitic disease in 2015, resulting in over 400,000 deaths due in large part to infection with Plasmodium falciparum (Pf) (WHO 2015). A vaccine capable of preventing malaria infection would prevent disease, death and onward parasite transmission and as such would constitute a critical tool in disease reduction as well as parasite eradication. However, all licensed vaccines to date have targeted viruses and bacteria, and although malaria vaccine development has seen some recent progress, a vaccine capable of delivering high levels of durable protection from Plasmodium parasites has yet to be developed.
During their complex life cycle within the mammalian host, malaria parasites present multiple targets for antibody-mediated interference with infection, providing a strong rationale that antibody-based vaccines could effectively interrupt the parasite transmission cycle and prevent disease and death. Whole attenuated parasite vaccine candidates and subunit vaccine candidates can both elicit protective antibody responses capable of neutralizing or destroying the parasite during infection.1,2,3,4,5,6,7,8 Attenuated parasites stimulate a broad antibody and T cell-mediated adaptive immune response against numerous parasite antigens. Subunit vaccines constitute a narrower approach where recombinant or vectored parasite antigen(s) are formulated with an immune-stimulatory adjuvant to elicit an antigen-specific response.
Antibodies can block Plasmodium parasite infection in the skin immediately after transmission, which occurs when an infected Anopheles mosquito injects tens to a few hundred sporozoite stages during a bite. Sporozoites are highly motile and traverse multiple cell types in search of a blood vessel, which gains them access to the blood circulation. In rodent models of malaria, it was shown that antibodies targeting the sporozoite can effectively prevent passage of the sporozoite to the liver by reducing the number of sporozoites ejected from the mosquito proboscis and also immobilizing the sporozoite in the dermis, thereby preventing their access to the circulation.9,10 Sporozoite motility and cell traversal are processes that require unique secreted and membrane-anchored proteins, which might be targeted with antibodies to prevent access to the blood circulation.11,12 Once in the circulation, sporozoites are rapidly transported to the liver where they again traverse multiple cell types as they cross the liver sinusoidal barrier and then infect a suitable hepatocyte. Leaving the circulation to enter the liver parenchyma also presents an opportunity for antibody-mediated prevention of infection as sporozoites are exposed to circulating antibodies that could target multiple sporozoite proteins involved in cell traversal and invasion, potentially preventing hepatocyte infection and in consequence, parasite replication in the liver. This in turn prevents the release of exo-erythrocytic merozoites and the establishment of a blood stage infection and its associated mortality and morbidity.13
The sporozoite and liver stages, collectively called the pre-erythrocytic (PE) stages of the parasite, are asymptomatic and are therefore attractive vaccine targets. Preventing the PE stages of infection will prevent all disease and the establishment of circulating sexual stage parasite populations that are transmissible. Indeed, endeavors are underway to identify new antibody targets for PE stages based on comprehensive sporozoite surface proteome and secretome data and a more in depth molecular understanding of the biological processes of parasite cell traversal and hepatocyte invasion.14,15 However, preclinical laboratory assays for assessing infection-inhibiting antibodies are extremely limited. Traditionally, rodent malaria models have been used for active immunization or for passive transfer of polyclonal or monoclonal antibodies followed by sporozoite challenge. However, there is significant evolutionary divergence between the malaria parasite species that infect rodents and humans, limiting the rodent malaria models as predictive preclinical experimental systems. Transgenic rodent malaria parasites carrying individual Pf antigens of interest have been created as bridging tools, but these are limited in the type and number of antigens that can be assessed.16,17,18 Thus, preclinical in vivo models that directly assess antibody efficacy against Pf challenge could be extremely helpful to inform which antigen candidates are to be advanced to clinical trials using controlled human malaria infection (CHMI).19 Without robust preclinical evidence for protective efficacy of subunit vaccine candidates against Pf challenge, CHMI trials have resulted in numerous vaccine candidate failures in the past.20,21,22,23,24,25 Although in vitro assays to evaluate antibody-mediated protection against Pf sporozoite infection have been established using monocultures of hepatoma cells or primary human hepatocytes26 they do not recapitulate the sporozoites infection route from the skin to the liver and therefore cannot capture antibody activities that block the parasite prior to hepatocyte infection.13,27,28
A bridge between preclinical in vitro assays and human CHMI trials could be built with liver-humanized mice, which fully support Pf PE infection.29,30 Measurement of parasite liver stage burden can then be used to assess passively transferred antibody efficacy.6,31,32 We have previously shown that these liver-chimeric humanized mice (referred to as FRG huHep mice), when challenged with a luciferase-expressing Pf parasite can be used to detect antibody-mediated reduction in liver infection by bioluminescent imaging.2,29,33,34 Here, we use FRG huHep mice to develop a robust platform to evaluate the protective efficacy of antibodies against natural Pf infection. We demonstrate that this model can differentiate monoclonal antibodies (mAbs) of varying infection-blocking activity and measure the infection-blocking activity of polyclonal immune IgG from human volunteers immunized with whole sporozoites. We also have adapted this model to measure antibody-mediated sterile protection using a physiologically relevant challenge dose. Importantly, mosquito bite challenge of FRG huHep mice discriminated antibody performance that was superior to in vitro assays, indicating the importance of using the natural Pf parasite infection route when assessing interventions against PE infection.
Results
Pf liver infection following mosquito bite challenge
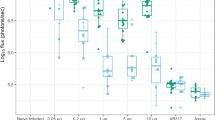
To determine antibody inhibitory function in vivo, we used the natural mosquito bite route of infection as opposed to intravenous (iv) injection of sporozoites, which has been used in the past but bypasses the skin stage and is less sensitive to antibody-mediated inhibition.33,35 However, mosquito bite infection is challenging to standardize, as it is difficult to control the precise sporozoite dose. Thus, we first asked if FRG huHep mouse infection with Pf parasites36 by mosquito bite yields a parasite stage liver burden of sufficient magnitude and consistency. We thus challenged FRG huHep mice with 25, 50, 75 or 100 Pf GFP_luc-infected mosquitoes each for 10 min. Liver stage burden was then quantified via bioluminescence on days 4–6 after infection (Fig. 1a). All mice had detectable liver infection on day 4 that increased on days 5 and 6 consistent with parasite liver stage growth over time (Fig. 1b). On day 6, all infections were statistically similar except the 25-bite group, which was significantly lower than both the 100 and 75 bite groups (p = 0.0021 and 0.0427, respectively, by two-way analysis of variance (ANOVA) for each day, Fig. 1b and Table 1). Given that 50 infected mosquito bites resulted in similar liver stage burden when compared to 75 and 100 mosquito bites (Table 1), we chose 50 bites as a standard dose for all further experiments. This dose proved to be consistent in terms of achieving a high magnitude of liver stage infection in all mice with a reasonable degree of variability that allowed us to detect antibody-mediated reductions in parasite liver infection in subsequent experiments (Supplementary Fig. S1).

FRG huHep mice are susceptible to P. falciparum infection by mosquito bite and parasite liver stage burden can be measured noninvasively by bioluminescent imaging. a Overall schematic of passive transfer and mosquito bite infection of FRG huHep mice. Mice are administered antibody 16–24 h prior to infection with Pf GFP_luc-infected mosquitoes followed by subsequent assessment of liver stage burden by bioluminescence at 4–6 days post infection. b Mice were exposed to the bites of 25, 50, 75 or 100 mosquitoes and tracked for liver stage burden from days 4–6. Individual mice are shown as data points with the mean for each group shown by the color-corresponding connecting line
Noninvasive quantitation of Pf parasite burden in the liver following passive antibody transfer
Bioluminescent imaging is a rapid and non-invasive method for assessing parasite liver stage burden. However, our initial experiments using mosquito bite infection showed variability within each experiment, which could preclude assessment of small changes in parasite burden such as those caused by moderate inhibition of infection with antibodies. Thus, we compared bioluminescent imaging and quantitative reverse transcriptase PCR (qRT-PCR) using parasite-specific probes to detect antibody-mediated reduction of liver infection. The latter method involves RNA extraction from infected liver and detection of parasite RNA by qRT-PCR.6,31 We compared these two methods by assessing the effect of passive transfer of 5, 50 and 150 μg of an anti-circumsporozoite protein (CSP) mAb on liver stage burden. A schematic example of sampling infected livers by both methods is shown in Fig. 2a. Six days after mosquito bite infection, liver stage burden was detectable in all mock-treated mice (mice receiving an equivalent dose of non-specific IgG) by both bioluminescent imaging and qRT-PCR (Fig. 2b) with considerable variability in both (coefficient of variance of 296 and 56% for qRT-PCR and bioluminescence, respectively). Both methods detected a significant reduction of liver stage burden following passive transfer of 150 μg of anti-CSP mAb with 3/5 mice having detectable liver burden by qRT-PCR and 5/5 by bioluminescent imaging (Fig. 2b). Whereas bioluminescent imaging could distinguish differences in liver stage parasite burden in mice given 5 and 50 μg of mAb, qRT-PCR could not, due to the large variability in signal (Fig. 2b). These results indicate that both bioluminescent imaging and this method of qRT-PCR detect relatively large differences in liver stage burden but that the former may be more useful in discriminating moderate reductions in liver stage burden.

Bioluminescent imaging is as accurate as qRT-PCR for assessment of liver stage burden. a Schematic representing liver sampling with qRT-PCR (upper panels) and bioluminescence (lower panels). Sampling of livers with biopsy punch for qRT-PCR is indicated by 12 circles where green circles indicate samples in which parasite RNA would be detected and red those which would be negative in mice with high liver burden (left) and low liver burden (right). In contrast, assessment of liver burden by bioluminescence is shown in the bottom panels and indicates a representative region of interest that can capture the entire liver. b FRG huHep mice were administered indicated doses of anti-CSP mAb 2A10 or 150 μg of non-specific murine IgG (mock). The following day, mice were infected with 50 Pf GFP_luc mosquito bites and assessed for liver burden at day 6 by bioluminescent imaging. Immediately following imaging, mice were sacrificed and 12 samples from each liver were collected and combined for RNA isolation and quantitation of liver burden by qRT-PCR. Liver burden by both qRT-PCR (left y-axis) and bioluminescence (right y-axis) are shown with each data point representing the mean liver burden from the 12 liver sections from one mouse for qRT-PCR and the total flux for each mouse measured by bioluminescence
FRG huHep mouse challenge to assess functional activity of mAbs
We next tested whether the FRG huHep/Pf GFP_luc challenge model could be used to measure differences in prevention of liver infection by distinct mAbs that target the same sporozoite antigen. To this end, we passively transferred three different mAbs of mouse origin that recognize the NANP repeat region of Pf CSP at 150 μg/mouse: mAb clone 2A10 (murine isotype IgG2b), mAb1 (murine isotype IgG1) and mAb2 (murine isotype IgG1). mAb 2A10 reduced liver infection to 51% of mock-treated mice while mAb1 and mAb2 reduced liver infection significantly to 10 and 31% of mock-treated mice, respectively (Fig. 3). To determine if the isotype of the mAb influenced antibody function, we also tested variants of mAb1 and mAb2 with isotypes known to better mediate Fc-dependent functions.37 Both mAb1-IgG3 and mAb2-IgG2a performed similarly to their IgG1isotype counterparts, indicating that antibody isotype was not a factor for in vivo performance in these experiments (Fig. 3). The results with these mAbs were consistent between replicate experiments (indicated by different colors in Fig. 3) despite differences in liver stage burden of mock groups (Supplementary Fig. S1). Importantly, the hierarchy in infection-inhibitory activity of each mAb, where mAb1 > mAb2> mAb 2A10, was not observed in an in vitro inhibition of sporozoite traversal and invasion (ISTI) assay where all mAbs performed similarly (Supplementary Fig. S2). This indicates that the assessment of antibody function in vitro may not fully reveal the inhibitory activity on sprozoite infection observed in vivo.

Monoclonal Abs of multiple Fc isotypes and species can be assessed in the FRG huHep/Pf GFP_luc challenge model. FRG huHep mice received 150 μg/mouse of non-specific IgG, mAb 2A10 or two novel anti-CSP mAb prior to mosquito bite infection and measurement of liver stage burden. Species and isotype are indicated for each mAb (e.g., mIgG1 for “murine IgG1” and “hu” for human). Data were collected over four independent experiments with 1–3 experiments/mAb and 4–5 mice/group/experiment. Each data point represents one mouse and mice within the same experiment are labeled in the same color. Bars indicate mean ± SEM with numbers above each bar indicating the mean % of mock for that group. Asterisks indicate statistical significance in one-way ANOVA comparing to mock-injected mice in the same independent experiments. Individual comparisons between mAb of the same specificity but different Fc isotypes/species are indicated with lines. ** is 0.01 > p > 0.001, **** is p < 0.0001
To increase the utility of the FRG huHep challenge model, we assessed whether mAbs of human origin can also be tested (i.e., derived from B cells from vaccinated volunteers). mAbs with human Fc regions set up a species mismatch between the antibody Fc region and the murine Fc receptor and this could diminish the contribution of Fc-dependent effector functions and obscure protection status.38 However, we observed that passive transfer of a version of mAb1 containing a human IgG3 (huIgG3) Fc region yielded similar results when compared to its murine counterparts (Fig. 3). Taken together, these data indicate that the FRG huHep/Pf GFP_luc challenge model discriminates distinct mAb blocking activities and enables functional assessment of mAbs of various Fc isotypes of both murine and human origin.
Inhibitory activity of polyclonal IgG from whole sporozoite-immunized volunteers
We next assessed the inhibitory activity of human polyclonal antibodies collected after immunization of volunteers with live-attenuated Pf sporozoites.
Immune serum or plasma from malaria vaccine trials is often limited and in previous studies using passive transfer of huIgG, the dose of IgG was dictated by sample availability.2,34 Thus, to first characterize the range of huIgG inhibitory activity in the FRG huHep/Pf GFP_luc challenge model, polyclonal huIgG pooled from samples collected from whole sporozoite-immunized volunteers2,34 was passively transferred at 0.5, 2, 4, 8 and 16 mg/mouse. We detected significant reductions in parasite liver stage burden as compared to mice receiving equivalent doses of pre-immune huIgG at doses as low as 4 mg/mouse (Fig. 4a). The serum levels of huIgG in mice receiving these inhibitory doses at the time of challenge were an average of 1.5, 4.5 and 8.3 mg/mL for doses of 4, 8 and 16 mg/mouse, respectively (Fig. 4b). Together, these data indicate that a range of huIgG concentrations can be used to determine antibody functionality in the FRG huHep/Pf GFP_luc challenge model and that a dose of 16 mg/mouse can closely recapitulate the concentration of huIgG observed in human serum.39

Human polyclonal IgG can be used over a wide dose range in the FRG huHep/Pf GFP_luc challenge model. FRG huHep mice (n = 4–5/group) received indicated doses of pooled pre-immune or immune polyclonal IgG at indicated doses. a Parasite liver stage burden was measured at day 6 post 50 mosquito bite challenge and the liver stage burden of each mouse receiving huIgG from immunized volunteers (Immune) was normalized to the average of the dose-matched huIgG from the pre-immunized volunteers (Pre-immune). Mean ± SEM is plotted with comparisons between pre-immune and immune liver stage burdens carried out by Mann–Whitney U test. Asterisks indicate a significant difference between means where * is p < 0.05 and ** is p ≤ 0.01. b Mice were bled immediately prior to challenge and serum was collected for measurement of circulating huIgG by ELISA. Concentration is depicted on the y-axis with mean ± SEM plotted for each group. Included are groups of mice that received a murine anti-CSP mAb or PBS (negative controls, n = 2 and 6, respectively)
We next used huIgG isolated from volunteers immunized with whole sporozoites and subsequently challenged by CHMI. The first set of huIgG was derived from volunteers intravenously immunized with four doses of 2.7 × 105 purified, cryopreserved irradiated sporozoites (PfSPZ).2 CHMI 6 months after final immunization demonstrated protection in 6/11 volunteers.2 We obtained sufficient sample to transfer 8 mg of huIgG/FRG huHep mouse (n = 3–10 mice/volunteer) taken at the time of CHMI from five protected and four non-protected volunteers. Control FRG huHep mice received 8 mg/mouse of pooled pre-immune huIgG from all nine volunteers and all mice were challenged by 50 Pf GFP_luc-infected mosquito bites. Compared to mice receiving pooled pre-immune huIgG, the immune huIgG from 4/5 protected volunteers significantly reduced parasite liver infection while immune huIgG from only 1/4 non-protected volunteers demonstrated significant reduction of liver infection (Fig. 5a). When grouped together, mice receiving immune huIgG from protected volunteers showed significantly reduced parasite liver burden (38% of pre-immune) as compared to mice receiving huIgG from non-protected volunteers, which only had a non-significant (66.6% of pre-immune) reduction in liver stage burden (Fig. 5b). However, this correlation was not evident on an individual volunteer basis as the mean parasite liver burden of mice receiving huIgG from a single volunteer (i.e., 1 mean/volunteer) was not statistically lower for individuals in the protected group as compared to the non-protected group despite a lower numerical value (Fig. 5c). Thus, antibody inhibitory function in vivo was not a clear correlate of protection for volunteers in this study. Regardless, the observed trends in differences between protected versus non-protected huIgG were not detected using an in vitro ISTI assay where IgG from 3/3 non-protected volunteers tested significantly inhibited sporozoite invasion whereas IgG from only 3/5 protected volunteers showed significant inhibition of invasion (Supplementary Fig. S3A, B). Comparing in vitro inhibition of either cell traversal or invasion to FRG huHep/Pf GFP_luc challenge data also yielded no correlation between the two assays suggesting a fundamental difference between sporozoite infection in the in vitro assay and the in vivo assay (Supplementary Fig. S3C ). Finally, the in vivo data included a replicate with a subset of immune huIgG to assess the reproducibility. We found results to be consistent between experiments using different batches of Pf GFP_luc-infected infected mosquitoes (Supplementary Fig. S4).

The FRG huHep/Pf GFP_luc challenge model can distinguish functional from non-functional antibodies from human volunteers. FRG huHep mice were administered 8 mg/mouse of IgG from volunteers 6 months after immunization with four doses of 2.7 × 105 irradiated sporozoites (PfSPZ) and immediately prior to challenge by infectious mosquito bite. Mice were then infected by bite of 50 Pf GFP_luc-infected mosquitos and liver stage burden assessed at day 6 post infection. Data shown are a combination of two independent experiments as delineated in Figure S4 and expressed as a percent of pre-immune parasite liver burden (% of pre-immune). a Liver stage burdens of mice (n = 3–10/volunteer) receiving post-immunization IgG were normalized to mice receiving pooled pre-immune IgG (n = 13). Data points are shown with bars as mean ± SEM for each volunteer grouped by protection status. Asterisks indicate significant differences from the pre-immune group as measured by one-way ANOVA with Kruskal–Wallis post-test. b Data in a arranged such that each mouse is plotted as a single data point and grouped according to their treatment, pre-immune huIgG, immune huIgG from a CHMI protected volunteer or immune huIgG from a non-protected CHMI volunteer. Above each group is the mean % of pre-immune as well as asterisks indicating a significant difference from the mean of the pre-immune group as measured by one-way ANOVA with Kruskal-Wallis post-test. An additional Mann–Whitney test comparing the means of protected and non-protected is indicated by bar and resulting p value. c Data represented as one mean per volunteer with Mann-Whitney test used to determine if the mean % of mock are different. For all comparisons ** is p ≤ 0.01, *** is p ≤ 0.001 and **** is p ≤ 0.0001
We next tested a set of huIgG from volunteers immunized by the chloroquine prophylaxis with sporozoites (CPS) protocol.40 CPS administers fully infectious sporozoites by mosquito bite to volunteers that concurrently receive chloroquine, which leads to complete liver stage development, release of exo-erythrocytic merozoites and subsequent elimination of the first cycle of blood stage parasites. CPS has proven to be highly efficacious,6,40,41 and in this trial protected 5/8 volunteers from CHMI. Due to sample limitations, we only transferred 5 mg huIgG into each FRG huHep mouse (3–5 mice/volunteer). Controls received either pre-immune huIgG or only phosphate-buffered saline (PBS). There was no significant difference between pre-immune huIgG and PBS-treated mice (Supplementary Fig. S5A). When compared to mice receiving pre-immune huIgG, immune huIgG from 1/5 protected volunteers significantly reduced parasite liver stage burden with another non-significantly reducing liver stage burden to 36% of control (Fig. 6a). In contrast, immune huIgG from none of the three non-protected volunteers reduced parasite liver burden (Fig. 6a). Grouping of mice based on those that received immune huIgG from either protected or non-protected volunteers, as in Fig. 5b, revealed that the parasite liver stage burden of mice receiving “protected” immune huIgG was lower (68% of PBS) than mice receiving either pre-immune huIgG (148% of PBS) or “non-protected” immune huIgG (160% of PBS, Fig. 6b). Grouping the mean parasite liver stage burdens of mice receiving huIgG from a single volunteer (one mean/volunteer) as in Fig. 5c revealed a lower average parasite liver stage burden after passive transfer of “protected” huIgG (72% of PBS) as compared to “non-protected” huIgG (166%, Fig. 6c). Again, these in vivo results were not predicted by the in vitro ISTI assay (Supplementary Fig. S5B). Although limited by small sample sizes, these data together demonstrate that the FRG huHep/Pf GFP_luc challenge model can be used to distinguish functional from non-functional polyclonal antibodies derived from volunteers vaccinated with whole sporozoites. The trends we observed also suggest that this model could be used to establish correlates of protection based on the in vivo function of antibodies but will likely require analysis of vaccine trials with larger cohort sizes.

Antibody function in the FRG huHep/Pf GFP_luc challenge model can be used as a correlate of protection. FRG huHep were administered 5 mg/mouse of IgG collected from volunteers either before (“Pre-Imm”) or 2 weeks after three immunizations with 15 Pf-infected mosquito bites under chloroquine prophylaxis. Mice were infected by the bite of 50 Pf GFP_luc-infected mosquitos and liver burden assessed at day 6 post infection. a Parasite liver stage burdens of mice (n = 3–5/volunteer) receiving pooled pre-immunization or individual post-immunization huIgG were normalized to mice receiving an equivalent volume of PBS (% of PBS). Data points are shown with bars as mean ± SEM for each volunteer and grouped by protection status. Asterisks indicate significant differences from the pre-immune group as measured by one-way ANOVA with Kruskal–Wallis post-test. b Data in a but arranged such that each mouse is plotted as a single data point and grouped by treatment with pre-immune huIgG, immune huIgG from a protected volunteer or immune huIgG from a non-protected volunteer. Above each group is the mean % of PBS as well as asterisks indicating a significant difference from the mean of pre-immune as measured by one-way ANOVA with Kruskal–Wallis post-test. An additional Mann–Whitney test comparing the means of protected and non-protected is indicated by bar and resulting p value. c Data represented as one mean per volunteer with Mann–Whitney test used to determine if the mean % of PBS between mice receiving protected and non-protected huIgG are different. For all comparisons * is p < 0.05 and ** is p ≤ 0.01
The ultimate goal of malaria vaccination is sterile protection, i.e., the complete prevention of parasite egress from the liver and subsequent onset of blood stage infection. Measuring reduction of liver stage burden as shown here does not directly assess sterile protection. We addressed this by performing passive mAb transfer followed by challenge with five Pf NF54-infected mosquito bites (the number of infected mosquitoes used for CHMI) and used the transition to blood stage infection instead of parasite liver burden as an endpoint of protection. At this low challenge dose, reductions in liver infection cannot be studied by bioluminescent imaging as the luminescence derived from this smaller infection is at the limit of detection without antibody treatment. Mice in this experiment were given 150, 50 or 15μg/mouse of mAb 2A10 the day prior to challenge and injected with human red blood cells on day 6.5 to allow parasite transition into the blood. The following day (day 7), peripheral blood was taken and parasite density measured by Plasmodium 18S rRNA qRT-PCR. None of the mice treated with non-specific mIgG were protected as all mice had detectable blood stage infection. However, in mice that received 150 μg mAb/mouse, a dose which reduced liver burden by 49% after 50 mosquito bite challenge, 2/5 were sterilely protected from the 5 bite challenge. Reduction in dose to 50 μg/mouse also resulted in 2/5 mice protected while an even lower dose of 15μg failed to show any protection (Fig. 7a). Parasite densitites in infected mice that received 150 μg of mAb were 52-fold lower than mIgG-treated mice (p < 0.05). Unprotected mice in the 50 μg dose group and the 15μg group had non-significant reductions in parasite densitites by 45-fold and 4.8-fold, respectively. We also determined the circulating mAb levels at the time of challenge in these mice to be an average of 38, 9.6 and 4.1 μg/mL in the 150, 50 and 15 μg/mouse groups, respectively. Thus, in addition to determining reductions in parasite liver burden following passive transfer, FRG-huHep mice can be used to measure sterile protection using a natural mosquito bite challenge that is equivalent to CHMI using a qRT-PCR endpoint assay also used in CHMI studies.34,42,43

The FRG huHep/Pf challenge model can be used to assess sterile protection. FRG huHep mice (n = 4–5 mice/group) were administered indicated dose of mAb 2A10 or non-specific mIgG 1 day prior to challenge by bite of five PfNF54-infected mosquitos. On day 6.5, mice were iv-injected with 400 μL of human red blood cells at 70% hematocrit. On day 7, peripheral blood was collected and used to assess presence of parasitemia by qRT-PCR. a Copies/mL of parasite 18S rRNA for each indicated dose of mAb 2A10 with negative mice plotted on the x-axis. Data points are individual mice with bars representing the mean ± SEM for each group. As a negative qRT-PCR control, mIgG-treated mice were bled prior to blood stage transition on day 6 (“150 μg mIgG D6”). Comparions between mIgG and 2A10-treated groups were carried out by one-way ANOVA and Kruskall–Wallis post-test. Significant differences are indicated by asterisk where p < 0.05. b Serum was collected immediately prior to challenge and circulating levels of mAb 2A10 were measured by ELISA. Each data point is one mouse with bars representing mean ± SEM shown for each group and numerical mean above each data set
Discussion
The malaria vaccine candidate RTS,S containing the imunodominant protein on the sporozoite surface, PfCSP, has undergone extensive clinical testing. It has achieved modest protection in phase III trials that correlate with anti-CSP-antibody titers and with CD4+ T cell responses3,4—evidence that antibodies against sporozoite antigens can afford protection against malaria in humans. This is corroborated by decades of studies in rodent malaria models using both active immunization and passive transfer of mAbs that demonstrate infection-blocking activity.9,33,35,44,45,46,47 Furthermore, clinical trials using immunization with live-attenuated sporozoites elicit anti-sporozoite antibodies that inhibit sporozoite infection in vitro and in vivo, and in some cases these antibody titers and in vitro function correlate with protection.1,2,6,48 Targeting the liver stages of parasite infection with T cells also affords sterile protection in rodent malaria models.49,50,51 However, the clinical performance of T cell based malaria vaccines has been low.52,53,54
RTS,S partial efficacy provided the impetus to embark on the development of a second generation subunit vaccine that not only boosts the quality, magnitude and durability of anti-CSP antibodies, but also incorporates additional antibody targets capable of blocking Pf sporozoite infection. However, these efforts are encumbered by the lack of preclinical models for natural Pf sporozoite infection delivered by mosquito bite. Here, we optimized a regimen of Pf infection by mosquito bite in FRG huHep mice that is consistent and robustly measures antibody efficacy with adequate group sizes over sequential experiments. This challenge strategy also proved sensitive enough to noninvasively measure reductions in parasite liver stage burden following passive transfer of mAbs in a manner that was at least as sensitive as qRT-PCR of infected liver tissue. Our noninvasive bioluminescent readout was superior to a qRT-PCR method that avoids processing the entire liver, but increased sensitivity might be achieved if whole liver RNA preparation and qRT-PCR were used.6,31,55 In addition, bioluminescent imaging is less time and reagent intensive as demonstrated by the fact that the parasite liver stage burden of 60 mice can be assessed in only approximately 1.5 hours by a single experienced researcher.
The FRG huHep/Pf GFP_luc challenge model can thus be used to down-select mAbs based on their in vivo function prior to clinical advancement of either the mAb or targeted antigen. Indeed, we were able to discriminate low-performing anti-CSP mAbs such as clone 2A10 from those reducing parasite liver stage burden by up to 90%. Importantly, antibody efficacies were maintained after switching to different mouse IgG subclasses or human Fc. This is critical as mAb with human Fc (i.e., those isolated from vaccinated volunteers) could set up a mismatch between the human antibody and the FRG huHep mouse host. This mismatch could obscure Fc-dependent antibody function given the poor performance of human Fc regions in mice.38 While we did not see an impact of the Fc region on Ab performance, we only tested mAbs recognizing CSP, and previous reports have also shown that the Fc region of anti-CSP antibodies is dispensable.44,56 Thus testing of non-CSP specific antibodies could show different results in the FRG huHep mouse if Fc-dependent mechanisms exist for other antigen targets. One such antibody targeting the α-gal glycan is heavily Fc-dependent in a rodent malaria model.57 As similar antibodies become available for Pf, it will be important to study their efficacy in FRG huHep mice. Nevertheless, the FRG huHep/Pf GFP_luc challenge model is sensitive, flexible and medium throughput, allowing for the functional activity screening of both mAb and polyclonal IgG.
The in vivo efficacy of antibodies against single antigens, such as CSP, can also be tested in standard mice using transgenic rodent malaria parasites where the endogenous antigen of interest has been replaced by the Pf ortholog.16,17,18 However, creation of these parasites is complex and the Pf protein must fully complement the cognate rodent malaria parasite protein to yield a viable parasite. This becomes increasingly difficult if combinations of antibodies targeting multiple proteins is desired as parasites carrying all Pf proteins of interest must be made—requiring multiple rounds of transgenesis and increasing the likelihood of non-viable parasites. This approach is also not suitable for use with polyclonal antibodies with unknown specificities such as those derived from whole sporozoite immunizations which might elicit thousands of different antigen specificities.58 Polyclonal huIgG isolated from volunteers immunized with whole sporozoites have been tested for the ability to prevent liver stage infection in human liver-chimeric mice2,6,34 but none of these studies rigorously investigated the range of huIgG dose needed to determine protection nor have they investigated antibody function in vivo as a correlate of protection. Here, we determined that a wide range of polyclonal huIgG could be used to observe antibody function and a dose of 16 mg/mouse is needed to recapitulate levels of circulating huIgG found in human serum. Knowing these requirements will help guide sampling schedules in clinical trials where determining the function of vaccine-elicited antibodies in vivo is desired.
We also investigated polyclonal huIgG activity elicited after whole sporozoite immunization in vivo as a correlate of protection. Using huIgG from volunteers 6 months after immunization with the irradiated PfSPZ vaccine, we found a weak correlation between antibody function and protection of volunteers after CHMI. We observed a greater number of protected volunteers whose huIgG mediated significant inhibition of liver stage infection in FRG huHep mice, but the correlation between the ability of an individual’s huIgG to reduce liver stage burden and protection was not significant—largely due to a single volunteer whose huIgG resulted, surprisingly, in an increased liver stage burden. Thus, we could not establish a clear, predictive correlate of protection in this study. This could be due to limitations in antibody potency, the small sample size or the fact that antibodies may play a minor role in protection in this particular study. Immunization with irradiated sporozoites such as PfSPZ has been shown in animal models to be heavily dependent on liver-resident cytotoxic T cells,59,60 thus establishing a dominant correlate of protection using only antibody function might be unlikely. Still, our data add to the evidence that vaccination with irradiated sporozoites induces long-lasting antibody responses capable of reducing the number of sporozoites reaching the liver and is in agreement with previous research.2
In a further set of experiments using polyclonal huIgG from volunteers immunized by CPS, we also observed a weak correlation between in vivo huIgG function and protection from CHMI despite less robust antibody function in individual volunteers. Sample sizes were again small, but huIgG from protected volunteers reduced parasite liver stage burden to a greater degree than non-protected volunteers regardless of how the data were analyzed. While conclusions regarding correlates of protection in both of these trials should be tempered, it is clear that we were able to detect differences in antibody function between volunteers and between groups of distinct protection status. This suggests that similar studies using larger clinical cohort sizes could lead to the generation of statistically significant results and could be a critical tool for identifying correlates or mechanisms of protection in multi-antigen or whole parasite vaccination trials. Even if larger studies reveal no correlates of protection with humoral immunity, knowing which arms of the immune system mediate protection can help guide vaccine design or delivery to best target that immune compartment.
Importantly, we also show that FRG-huHep mice can be used to assess sterile protection when a highly sensitive Plasmodium 18 s rRNA qRT-PCR assay is used to detect blood stage infection after a lower, five mosquito bite challenge dose. This is the same challenge dose used in CHMI trials and is the same diagnostic endpoint assay used in such studies.19,34,42,43 All mock-treated mice in our sterile protection experiment became blood stage positive at day 7. Sterile protection is a binary readout with a limited dynamic range. However, while sterile protection was observed at the two highest doses of mAb 2A10 there was none in the lowest dose group, suggesting that this model is still sensitive to antibody dose and is reflective of overall efficacy. When coupled with the ability to modulate circulating mAb levels as shown here, it will be possible to determine the concentrations of antibody required to achieve sterile protection. This will be useful for screening and down-selection of antibodies against novel antigens or epitopes and the pre-CHMI determination of protective levels of antibodies that ought to be achieved by active immunization.
Finally, a key finding across all of our experiments was that the in vitro ISTI assay did not predict performance in the FRG huHep/Pf GFP_luc challenge model. The mAbs we tested all performed equally well in vitro yet exhibited large differences in reductions of parasite liver stage burden in vivo. In vitro results for clinical samples also did not predict any trends in inhibition and in fact over-estimated antibody function in non-protected volunteers. Thus, in vitro assays should be interpreted with caution and not used in isolation for down-selection of antibodies/targets, nor should they be used to rank antibody efficacy. It is perhaps not surprising that antibody function in vitro does not accurately predict function in vivo as the sporozoite uses unique mechanisms to travel through the dermis, into the circulation, across the liver sinusoidal barrier and finally into the three-dimensional architecture of the liver for liver stage development.9,10,33,35,47 All current in vitro assays employ infection of hepatocytes in monoculture which is not representative of the sporozoites’ complex journey to the liver or the architecture of the target organ.26
FRG huHep mice still suffer some limitations for use in antibody studies as they lack B, T, NK and NKT cells. This precludes the detection of antibody efficacy if it relies on interactions between Fc regions and the Fc-receptors (FcR) on these cell types, although to our knowledge, there is no demonstration of antibody function against sporozoites being dependent on interacting with these cell types. Nevertheless, FRG huHep mice do harbor antigen-presenting cells such as dendritic cells and macrophages which could phagocytose antibody-bound sporozoites.61 Polymorphonuclear cells are also present in these mice62 and could augment antibody-mediated prevention of liver infection.57 However, all bone marrow-derived cells will contain mouse FcR that bind poorly to human Fc regions and may limit the function of transferred human mAb or huIgG.38 While the role of complement in protection against sporozoite infection is unclear,44,56,57 it is also unclear if these mice can fix complement via the classical/antibody-mediated pathway with either mouse or human antibodies. FRG huHep mice do make human C3 (data not shown), but it remains to be seen if this is functional in the classical complement pathway.
Despite their limitations, our data show that FRG huHep mice are a highly relevant laboratory model to study the role of inhibitory antibodies against natural Pf infection. mAbs against novel epitopes of existing antigen candidates or completely novel candidates can be screened in FRG huHep mice and will thereby help determine those that function best in vivo. The data can then help guide rational structure-based antigen design as well as down-select antigen candidates to move into CHMI trials using either passive immunization with mAbs or active immunization with the target antigen. Human liver-chimeric mice are also suitable models to test the in vivo function of polyclonal antibodies with diverse or unknown specificities such as those derived from vaccination with complex immunogens, including whole sporozoites. Testing of these antibodies in vivo will help identify correlates or contributing mechanisms of protection that cannot be discerned in vitro. Thus, the model described here can further enable rational optimization of protective malaria vaccine candidates and as such will form a bridge to clinical malaria vaccine development efforts.
Materials and methods
FRG huHep mouse challenges
Human hepatocyte donor-matched FRG huHep mice (both male and female, >4 months of age) were purchased from Yecuris, Inc. Repopulation of human hepatocytes was confirmed by serum albumin levels, and only animals with serum albumin levels >3 mg/mL were used. For passive transfer, mice were intravenously or intraperitoneally injected with indicated dose of anti-Pf CSP monoclonal antibody or huIgG 16–24 h prior to challenge. mAbs were kindly provided by PATH Malaria Vaccine Initiative where mAb1 is clone 3C1 and mAb2 is clone 2H8. For mosquito bite challenge, Anopheles mosquitos infected with Pf expressing GFP-luciferase or wild type NF54 were generated as previously described.36 Mosquito infection was quantified by midgut dissection at day 7–10 post-blood meal and were used only if >50% of mosquitos contained an average of >10 oocysts/midgut. All qualifying mosquitos were then pooled and re-distributed into cages with ~50 mosquitos/mouse with up to 250 mosquitos. For five-mosquito bite challenge, the number of mosquitos/mouse was adjusted to reflect infection prevalence (e.g., if 90% of mosquitos were infected, a total of 28 mosquitos were added to a cage to infect five mice). Mice, in groups of up to five, were then anesthetized with isoflurane and placed on a mesh screen covering the container of mosquitos while under isoflurane anesthesia via nose cone. Mosquitos were then allowed to feed for 10 min with lifting of mice every minute to encourage probing and injection of sporozoites rather than blood feeding. After 10 min mice were returned to normal activity. At day 6 post infection (peak of liver stage burden), mice were imaged for liver stage burden using bioluminescence and IVIS imaging as previously described.33,63 Briefly, mice were intraperitoneally injected with 100 μL of Rediject D-luciferin (Perkin Elmer) and imaged after 5 min for a five-minute exposure. Liver stage burden was assessed by placing an identical region of interest around the liver of each mouse and measuring total flux in pixels/second (p/s). Liver stage burden of all mice was normalized by setting the mean of the negative control group that received non-specific, species-matched IgG or pre-immune huIgG to 100% within each bite experiment. Liver stage burden was alternatively assessed in a subset of experiments by Plasmodium 18S rRNA RT-PCR normalized to hApoA1 mRNA, as previously reported.6,64
For assessment of sterile protection, mice were iv-injected with 400 μL of human red blood cells at 70% hematocrit in RPMI 6.5 days post challenge. On day 7, mice were bled via the retroorbital plexus and exactly 50 μL of blood was placed into 1 mL of Nuclisens EasyMag buffer (bioMérieux) and stored at −80 °C until extraction. Total RNA was extracted using an EasyMag instrument (bioMérieux) as described.43 Plasmodium 18S rRNA qRT-PCR was performed as described65 although newly described pan-Plasmodium 18S rRNA-specific reagents were used herein.66 Mice were considered positive if qRT-PCR was above the undetectable qRT-PCR signal obtained on day 6 post challenge.
ELISA for serum mAb concentration
Mice were bled via retroorbital plexus immediately prior to challenge and blood allowed to clot in BD serum separator tubes for 2 h. Serum was separated by centrifugation at 14,000×g for 2 min. For ELISA, Costar EasyWash (Corning) were coated with 2 μg/mL in coating buffer as previously described.34 Plates were then blocked with dilution/blocking buffer (0.05% Tween-20, 6% bovine serum albumin in PBS) for 1 h at room temperature. After washing, 50 μL of serum samples were applied in duplicate at 1:160 and 1:320 dilutions in dilution/blocking buffer. A standard curve was generated using eight two-fold dilutions of 2A10 starting at 625 ng/mL in dilution/blocking buffer and 50 μL applied in duplicate with samples. Standards and serum samples were incubated at room temperature for 2 h. After washing, HRP-conjugated anti-mouse IgG was applied at a 1:5000 dilution for 1 h at room temperature. Plates were developed with SureBlue™ TMB reagent for 4 min and stopped with SureBlue™ Stop reagent before reading absorbance at 450 nm. Serum concentrations were interpolated using a 4-parameter non-linear regression of the standard curve.
Ethics statement
All animal procedures were conducted in accordance with and approved by the Center for Infectious Disease Research Institutional Animal Care and Use Committee (IACUC) under protocol SK-16. The Seattle Biomed IACUC adheres to the NIH Office of Laboratory Animal Welfare standards (OLAW welfare assurance # A3640-01).
Preparation of monoclonal and polyclonal antibodies
Monoclonal antibody 1 (mAb 1, clone 3C1) was prepared as previously described.2 Monoclonal antibody 2 (mAb 2, clone 2H8) was generated under a Gennova-PATH Malaria Vaccine Initiative collaborative program by immunization of mice against full length PfCSP produced by Gennova. Hybridoma clone 2A10 specific for Pf CSP was obtained from MR4 and ProMab Biotechnologies, Inc. performed antibody production and purification. For IgG isolated from human serum, samples were prepared as recently published.2 Briefly, serum was used for extraction of IgG using protein G columns (GE Healthcare Life Sciences) following manufacturer’s protocol and concentrated using Amicon Ultra-15 centrifugal units (EMD Millipore) to 20–25 mg/mL in PBS.
In vitro inhibition of sporozoite invasion and traversal assay (ISTI)
Antibodies and IgG were tested at indicated concentrations in ISTI following previously published methods.67,68 Briefly, freshly dissected Pf sporozoites were incubated with antibodies at indicated concentrations in DMEM media containing 10% heat-inactivated FBS supplemented with glutamine, penicillin/streptomycin, Fungizone and FITC-dextran for 15 min a 37 °C. Sporozoites and media were then added to HC04 cells (ATCC, Inc.) plated 1 day prior at 105 cells/well in a 96-well plate at an MOI of 0.3 (sporozoites:HC04 cells). Plates were then spun at 500×g for 3 min and incubated for 90 min at 37 °C. Cells were then washed, trypsinized and transferred to a new 96-well plate where they were fixed and permeabilized using BD cytofix/cytoperm (BD Biosciences). Cells were then stained with anti-CSP mAb conjugated to AlexaFluor-647 and analyzed by flow cytometry using a BD LSRII and FloJo analysis software. Cells were considered “invaded” if they were CSP-positive and “traversed” if they had taken up FITC-dextran due to cell membrane wounding. Antibody-treated wells were normalized to either species-matched non-specific IgG for mAbs and to pooled pre-immune IgG at equivalent concentrations. Anti-CSP mAb 2A10 was included at 10 μg/mL in each assay as a positive control for inhibition.
Statistical analysis
Relevant statistical tests are indicated in figure legends and were conducted using GraphPad Prism 6.0 for Mac. Significant results are indicated in figures with relevant p values indicated by “*” in each figure legend. Comparisons not indicated in figures were non-significant with p > 0.05.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Seder, R. A., Chang, L. J., Enama, M. E., Zephir, K. L., Sarwar, U. N. & Gordon, I. J. et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365 (2013).
Ishizuka, A. S., Lyke, K. E., DeZure, A., Berry, A. A., Richie, T. L. & Mendoza, F. H. et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat. Med. 22, 614–623 (2016).
Kester, K. E., Cummings, J. F., Ofori-Anyinam, O., Ockenhouse, C. F., Krzych, U. & Moris, P. et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J. Infect. Dis. 200, 337–346 (2009).
Olotu, A., Fegan, G., Wambua, J., Nyangweso, G., Awuondo, K. O. & Leach, A. et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N. Eng. J. Med. 368, 1111–1120 (2013).
White, M. T., Bejon, P., Olotu, A., Griffin, J. T., Riley, E. M. & Kester, K. E. et al. The relationship between RTS,S vaccine-induced antibodies, CD4(+) T cell responses and protection against Plasmodium falciparum infection. PLoS One 8, e61395 (2013).
Behet, M. C., Foquet, L., van Gemert, G. J., Bijker, E. M., Meuleman, P. & Leroux-Roels, G. et al. Sporozoite immunization of human volunteers under chemoprophylaxis induces functional antibodies against pre-erythrocytic stages of Plasmodium falciparum. Malar. J. 13, 136 (2014).
Epstein, J. E., Paolino, K. M., Richie, T. L., Sedegah, M., Singer, A. & Ruben, A. J. et al. Protection against Plasmodium falciparum malaria by PfSPZ Vaccine. JCI Insight 2, e89154 (2017).
Lyke, K. E., Ishizuka, A. S., Berry, A. A., Chakravarty, S., DeZure, A. & Enama, M. E. et al. Attenuated PfSPZ vaccine induces strain-transcending T cells and durable protection against heterologous controlled human malaria infection. Proc. Natl. Acad. Sci. USA 114, 2711–2716 (2017).
Vanderberg, J. P. & Frevert, U. Intravital microscopy demonstrating antibody-mediated immobilisation of Plasmodium berghei sporozoites injected into skin by mosquitoes. Int. J. Parasitol. 34, 991–996 (2004).
Kebaier, C., Voza, T. & Vanderberg, J. Kinetics of mosquito-injected Plasmodium sporozoites in mice: fewer sporozoites are injected into sporozoite-immunized mice. PLoS Pathog. 5, e1000399 (2009).
Sinnis, P. & Zavala, F. The skin stage of malaria infection: biology and relevance to the malaria vaccine effort. Future Microbiol. 3, 275–278 (2008).
Sinnis, P. & Zavala, F. The skin: where malaria infection and the host immune response begin. Semin. Immunopathol. 34, 787–792 (2012).
Cha, S. J., Kim, M. S., Pandey, A. & Jacobs-Lorena, M. Identification of GAPDH on the surface of Plasmodium sporozoites as a new candidate for targeting malaria liver invasion. J. Exp. Med. 213, 2099–2112 (2016).
Lindner, S. E., Swearingen, K. E., Harupa, A., Vaughan, A. M., Sinnis, P. & Moritz, R. L. et al. Total and putative surface proteomics of malaria parasite salivary gland sporozoites. Mol. Cell. Proteomics 12, 1127–1143 (2013).
Swearingen, K. E., Lindner, S. E., Shi, L., Shears, M. J., Harupa, A. & Hopp, C. S. et al. Interrogating the Plasmodium sporozoite surface: identification of surface-exposed proteins and demonstration of glycosylation on CSP and TRAP by mass spectrometry-based proteomics. PLoS Pathog. 12, e1005606 (2016).
Espinosa, D. A., Yadava, A., Angov, E., Maurizio, P. L., Ockenhouse, C. F. & Zavala, F. Development of a chimeric Plasmodium berghei strain expressing the repeat region of the P. vivax circumsporozoite protein for in vivo evaluation of vaccine efficacy. Infect. Immun. 81, 2882–2887 (2013).
Bauza, K., Malinauskas, T., Pfander, C., Anar, B., Jones, E. Y. & Billker, O. et al. Efficacy of a Plasmodium vivax malaria vaccine using ChAd63 and modified vaccinia Ankara expressing thrombospondin-related anonymous protein as assessed with transgenic Plasmodium berghei parasites. Infect. Immun. 82, 1277–1286 (2014).
Persson, C., Oliveira, G. A., Sultan, A. A., Bhanot, P., Nussenzweig, V. & Nardin, E. Cutting edge: a new tool to evaluate human pre-erythrocytic malaria vaccines: rodent parasites bearing a hybrid Plasmodium falciparum circumsporozoite protein. J. Immunol. 169, 6681–6685 (2002).
Sauerwein, R. W., Roestenberg, M. & Moorthy, V. S. Experimental human challenge infections can accelerate clinical malaria vaccine development. Nat. Rev. Immunol. 11, 57–64 (2011).
Cummings, J. F., Spring, M. D., Schwenk, R. J., Ockenhouse, C. F., Kester, K. E. & Polhemus, M. E. et al. Recombinant Liver Stage Antigen-1 (LSA-1) formulated with AS01 or AS02 is safe, elicits high titer antibody and induces IFN-gamma/IL-2 CD4+ T cells but does not protect against experimental Plasmodium falciparum infection. Vaccine 28, 5135–5144 (2010).
Kester, K. E., Gray Heppner, D. Jr., Moris, P., Ofori-Anyinam, O., Krzych, U. & Tornieporth, N. et al. Sequential Phase 1 and Phase 2 randomized, controlled trials of the safety, immunogenicity and efficacy of combined pre-erythrocytic vaccine antigens RTS,S and TRAP formulated with AS02 Adjuvant System in healthy, malaria naive adults. Vaccine 32, 6683–6691 (2014).
McConkey, S. J., Reece, W. H., Moorthy, V. S., Webster, D., Dunachie, S. & Butcher, G. et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 9, 729–735 (2003).
Ockenhouse, C. F., Sun, P. F., Lanar, D. E., Wellde, B. T., Hall, B. T. & Kester, K. et al. Phase I/IIa safety, immunogenicity, and efficacy trial of NYVAC-Pf7, a pox-vectored, multiantigen, multistage vaccine candidate for Plasmodium falciparum malaria. J. Infect. Dis. 177, 1664–1673 (1998).
Porter, D. W., Thompson, F. M., Berthoud, T. K., Hutchings, C. L., Andrews, L. & Biswas, S. et al. A human Phase I/IIa malaria challenge trial of a polyprotein malaria vaccine. Vaccine 29, 7514–7522 (2011).
Richie, T. L., Charoenvit, Y., Wang, R., Epstein, J. E., Hedstrom, R. C. & Kumar, S. et al. Clinical trial in healthy malaria-naive adults to evaluate the safety, tolerability, immunogenicity and efficacy of MuStDO5, a five-gene, sporozoite/hepatic stage Plasmodium falciparum DNA vaccine combined with escalating dose human GM-CSF DNA. Hum. Vaccin. Immunother. 8, 1564–1584 (2012).
Steel, R. W., Kappe, S. H. & Sack, B. K. An expanding toolkit for preclinical pre-erythrocytic malaria vaccine development: bridging traditional mouse malaria models and human trials. Future Microbiol. 11, 1563–1579 (2016).
Amino, R., Giovannini, D., Thiberge, S., Gueirard, P., Boisson, B. & Dubremetz, J. F. et al. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe 3, 88–96 (2008).
Tavares, J., Formaglio, P., Thiberge, S., Mordelet, E., Van Rooijen, N. & Medvinsky, A. et al. Role of host cell traversal by the malaria sporozoite during liver infection. J. Exp. Med. 210, 905–915 (2013).
Vaughan, A. M., Mikolajczak, S. A., Wilson, E. M., Grompe, M., Kaushansky, A. & Camargo, N. et al. Complete Plasmodium falciparum liver-stage development in liver-chimeric mice. J. Clin. Invest. 122, 3618–3628 (2012).
Mikolajczak, S. A., Vaughan, A. M., Kangwanrangsan, N., Roobsoong, W., Fishbaugher, M. & Yimamnuaychok, N. et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535 (2015).
Foquet, L., Hermsen, C. C., van Gemert, G. J., Van Braeckel, E., Weening, K. E. & Sauerwein, R. et al. Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection. J. Clin. Invest. 124, 140–144 (2014).
Foquet, L., Meuleman, P., Hermsen, C. C., Sauerwein, R. & Leroux-Roels, G. Assessment of parasite liver-stage burden in human-liver chimeric mice. Methods Mol. Biol. 1325, 59–68 (2015).
Sack, B. K., Miller, J. L., Vaughan, A. M., Douglass, A., Kaushansky, A. & Mikolajczak, S. et al. Model for in vivo assessment of humoral protection against malaria sporozoite challenge by passive transfer of monoclonal antibodies and immune serum. Infect. Immun. 82, 808–817 (2014).
Kublin, J. G., Mikolajczak, S. A., Sack, B. K., Fishbaugher, M. E., Seilie, A. & Shelton, L., et al. Complete attenuation of genetically engineered Plasmodium falciparum sporozoites in human subjects. Sci. Transl. Med. 9, 371 (2017).
Keitany, G. J., Sack, B., Smithers, H., Chen, L., Jang, I. K. & Sebastian, L. et al. Immunization of mice with live-attenuated late liver stage-arresting Plasmodium yoelii parasites generates protective antibody responses to preerythrocytic stages of malaria. Infect. Immun. 82, 5143–5153 (2014).
Vaughan, A. M., Mikolajczak, S. A., Camargo, N., Lakshmanan, V., Kennedy, M. & Lindner, S. E. et al. A transgenic Plasmodium falciparum NF54 strain that expresses GFP-luciferase throughout the parasite life cycle. Mol. Biochem. Parasitol. 186, 143–147 (2012).
Kipps, T. J., Parham, P., Punt, J. & Herzenberg, L. A. Importance of immunoglobulin isotype in human antibody-dependent, cell-mediated cytotoxicity directed by murine monoclonal antibodies. J. Exp. Med. 161, 1–17 (1985).
Overdijk, M. B., Verploegen, S., Ortiz Buijsse, A., Vink, T., Leusen, J. H. & Bleeker, W. K. et al. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 189, 3430–3438 (2012).
Gonzalez-Quintela, A., Alende, R., Gude, F., Campos, J., Rey, J. & Meijide, L. M. et al. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin. Exp. Immunol. 151, 42–50 (2008).
Roestenberg, M., McCall, M., Hopman, J., Wiersma, J., Luty, A. J. & van Gemert, G. J. et al. Protection against a malaria challenge by sporozoite inoculation. N. Eng. J. Med. 361, 468–477 (2009).
Roestenberg, M., Teirlinck, A. C., McCall, M. B., Teelen, K., Makamdop, K. N. & Wiersma, J. et al. Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet 377, 1770–1776 (2011).
Hodgson, S. H., Douglas, A. D., Edwards, N. J., Kimani, D., Elias, S. C. & Chang, M. et al. Increased sample volume and use of quantitative reverse-transcription PCR can improve prediction of liver-to-blood inoculum size in controlled human malaria infection studies. Malar. J. 14, 33 (2015).
Murphy, S. C., Prentice, J. L., Williamson, K., Wallis, C. K., Fang, F. C. & Fried, M. et al. Real-time quantitative reverse transcription PCR for monitoring of blood-stage Plasmodium falciparum infections in malaria human challenge trials. Am. J. Trop. Med. Hyg. 86, 383–394 (2012).
Potocnjak, P., Yoshida, N., Nussenzweig, R. S. & Nussenzweig, V. Monovalent fragments (Fab) of monoclonal antibodies to a sporozoite surface antigen (Pb44) protect mice against malarial infection. J. Exp. Med. 151, 1504–1513 (1980).
Charoenvit, Y., Sedegah, M., Yuan, L. F., Gross, M., Cole, C. & Bechara, R. et al. Active and passive immunization against Plasmodium yoelii sporozoites. Bull. World Health Organ. 68, Suppl:26–Suppl:32 (1990).
Charoenvit, Y., Mellouk, S., Cole, C., Bechara, R., Leef, M. F. & Sedegah, M. et al. Monoclonal, but not polyclonal, antibodies protect against Plasmodium yoelii sporozoites. J. Immunol. 146, 1020–1025 (1991).
Vanderberg, J., Mueller, A. K., Heiss, K., Goetz, K., Matuschewski, K. & Deckert, M. et al. Assessment of antibody protection against malaria sporozoites must be done by mosquito injection of sporozoites. Am. J. Pathol. 171, 1405–1406 (2007). author reply 6.
Bijker, E. M., Bastiaens, G. J., Teirlinck, A. C., van Gemert, G. J., Graumans, W. & van de Vegte-Bolmer, M. et al. Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proc Natl Acad Sci USA 110, 7862–7867 (2013).
Bijker, E. M., Teirlinck, A. C., Schats, R., van Gemert, G. J., van de Vegte-Bolmer, M. & van Lieshout, L. et al. Cytotoxic markers associate with protection against malaria in human volunteers immunized with Plasmodium falciparum sporozoites. J. Infect. Dis. 210, 1605–1615 (2014).
Epstein, J. E., Tewari, K., Lyke, K. E., Sim, B. K., Billingsley, P. F. & Laurens, M. B. et al. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science 334, 475–480 (2011).
Doll, K. L. & Harty, J. T. Correlates of protective immunity following whole sporozoite vaccination against malaria. Immunol. Res. 59, 166–176 (2014).
Sheehy, S. H., Duncan, C. J., Elias, S. C., Choudhary, P., Biswas, S. & Halstead, F. D. et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol. Ther. 20, 2355–2368 (2012).
Hill, A. V., Reyes-Sandoval, A., O’Hara, G., Ewer, K., Lawrie, A. & Goodman, A. et al. Prime-boost vectored malaria vaccines: progress and prospects. Hum. Vaccin. 6, 78–83 (2010).
Hodgson, S. H., Ewer, K. J., Bliss, C. M., Edwards, N. J., Rampling, T. & Anagnostou, N. A. et al. Evaluation of the efficacy of ChAd63-MVA vectored vaccines expressing circumsporozoite protein and ME-TRAP against controlled human malaria infection in malaria-naive individuals. J. Infect. Dis. 211, 1076–1086 (2015).
Bruna-Romero, O., Hafalla, J. C., Gonzalez-Aseguinolaza, G., Sano, G., Tsuji, M. & Zavala, F. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. Int. J. Parasitol. 31, 1499–1502 (2001).
Ak, M., Bower, J. H., Hoffman, S. L., Sedegah, M., Lees, A. & Carter, M. et al. Monoclonal antibodies of three different immunoglobulin G isotypes produced by immunization with a synthetic peptide or native protein protect mice against challenge with Plasmodium yoelii sporozoites. Infect. Immun. 61, 2493–2497 (1993).
Yilmaz, B., Portugal, S., Tran, T. M., Gozzelino, R., Ramos, S. & Gomes, J. et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell. 159, 1277–1289 (2014).
Felgner, P. L., Roestenberg, M., Liang, L., Hung, C., Jain, A. & Pablo, J. et al. Pre-erythrocytic antibody profiles induced by controlled human malaria infections in healthy volunteers under chloroquine prophylaxis. Sci. Rep. 3, 3549 (2013).
Weiss, W. R., Sedegah, M., Beaudoin, R. L., Miller, L. H. & Good, M. F. CD8+ T cells (cytotoxic/suppressors) are required for protection in mice immunized with malaria sporozoites. Proc. Natl. Acad. Sci. USA 85, 573–576 (1988).
Rodrigues, M., Nussenzweig, R. S. & Zavala, F. The relative contribution of antibodies, CD4+ and CD8+ T cells to sporozoite-induced protection against malaria. Immunology 80, 1–5 (1993).
Celada, A., Cruchaud, A. & Perrin, L. H. Opsonic activity of human immune serum on in vitro phagocytosis of Plasmodium falciparum infected red blood cells by monocytes. Clin. Exp. Immunol. 47, 635–644 (1982).
Mazurier, F., Fontanellas, A., Salesse, S., Taine, L., Landriau, S. & Moreau-Gaudry, F. et al. A novel immunodeficient mouse model—RAG2 x common cytokine receptor gamma chain double mutants--requiring exogenous cytokine administration for human hematopoietic stem cell engraftment. J. Interferon Cytokine Res. 19, 533–541 (1999).
Miller, J. L., Murray, S., Vaughan, A. M., Harupa, A., Sack, B. & Baldwin, M. et al. Quantitative bioluminescent imaging of pre-erythrocytic malaria parasite infection using luciferase-expressing Plasmodium yoelii. PLoS One 8, e60820 (2013).
Foquet, L., Hermsen, C. C., van Gemert, G. J., Van Braeckel, E., Weening, K. E. & Sauerwein, R., et al. Vaccine-induced monoclonal antibodies targetingcircumsporozoite protein prevent Plasmodium falciparum infection. J. Clin. Invest. 124, 140–144 (2013)
Murphy, S. C., Daza, G., Chang, M. & Coombs, R. Laser cutting eliminates nucleic acid cross-contamination in dried-blood-spot processing. J. Clin. Microbiol. 50, 4128–4130 (2012).
Billman, Z. P., Seilie, A. M. & Murphy, S. C. Purification of Plasmodium sporozoites enhances parasite-specific CD8+ T cell responses. Infect. Immun. 84, 2233–2242 (2016).
Kaushansky, A., Rezakhani, N., Mann, H. & Kappe, S. H. Development of a quantitative flow cytometry-based assay to assess infection by Plasmodium falciparum sporozoites. Mol. Biochem. Parasitol. 183, 100–103 (2012).
Douglass, A. N., Metzger, P. G., Kappe, S. H. & Kaushansky, A. Flow cytometry-based assessment of antibody function against malaria pre-erythrocytic infection. Methods Mol. Biol. 1325, 49–58 (2015).
Acknowledgements
We would like to thank Moriya Tsuji (ADARC/Rockefeller University) for providing mAb 3C1 and the PATH Malaria Vaccine Initiative/Gennova for providing mAb 2H8 as well as the 3C1 and 2H8 isotype variants. We would also like to thank the Center for Infectious Disease Research vivarium staff for their expertise in animal care and Yecuris, Inc. for the coordination of animal production and delivery. This work was funded by the Bill and Melinda Gates foundation (Investment ID: 24922), MVI/PATH (grant # GAT.0888-30-01259780-COL) and NIH grant # F32 AI 114113.
Author information
Authors and Affiliations
Contributions
B.K.S. designed, conducted and analyzed data for all experiments. S.A.M. assisted in experimental design, data analysis and mosquito bite infections. M.F. assisted in experimental design, Pf parasite production and mosquito bite infection. A.M.V. assisted in experiment design and design of mosquito bite infection. E.F., T.N., W.B., and M.J.N. produced infected mosquitos and coordination of infection. L.F. assisted in experiment design and passive transfer studies. R.W.J.S. assisted in design and conducted experiments involving ISTI with mAb. M.B., I.R., J.W., A.S., A.I., B.F., R.A.S., R.W.S., S.H., S.C. and K.S. assisted in experimental design, coordination and data analysis of clinical samples. Z.P.B. and S.C.M. performed whole blood qRT-PCR assays. S.H.I.K. designed experiments, assisted in data analysis and coordination of all experiments. All authors have participated in the drafting and revising of the manuscript and have approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sack, B.K., Mikolajczak, S.A., Fishbaugher, M. et al. Humoral protection against mosquito bite-transmitted Plasmodium falciparum infection in humanized mice. npj Vaccines 2, 27 (2017). https://doi.org/10.1038/s41541-017-0028-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-017-0028-2
This article is cited by
-
Establishing RTS,S/AS01 as a benchmark for comparison to next-generation malaria vaccines in a mouse model
npj Vaccines (2024)
-
Comparative analyses of functional antibody-mediated inhibition with anti-circumsporozoite monoclonal antibodies against transgenic Plasmodium berghei
Malaria Journal (2023)
-
Evaluating the stability of host-reference gene expression and simultaneously quantifying parasite burden and host immune responses in murine malaria
Scientific Reports (2023)
-
Anti-TRAP/SSP2 monoclonal antibodies can inhibit sporozoite infection and may enhance protection of anti-CSP monoclonal antibodies
npj Vaccines (2022)
-
Creation and preclinical evaluation of genetically attenuated malaria parasites arresting growth late in the liver
npj Vaccines (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}