
Abstract
Delusions are a difficult-to-treat and intellectually fascinating aspect of many psychiatric illnesses. Although scientific progress on this complex topic has been challenging, some recent advances focus on dysfunction in neural circuits, specifically in those involving dopaminergic and glutamatergic neurotransmission. Here we review the role of cholinergic neurotransmission in delusions, with a focus on nicotinic receptors, which are known to play a part in some illnesses where these symptoms appear, including delirium, schizophrenia spectrum disorders, bipolar disorder, Parkinson, Huntington, and Alzheimer diseases. Beginning with what we know about the emergence of delusions in these illnesses, we advance a hypothesis of cholinergic disturbance in the dorsal striatum where nicotinic receptors are operative. Striosomes are proposed to play a central role in the formation of delusions. This hypothesis is consistent with our current knowledge about the mechanism of action of cholinergic drugs and with our abstract models of basic cognitive mechanisms at the molecular and circuit levels. We conclude by pointing out the need for further research both at the clinical and translational levels.
Similar content being viewed by others
Introduction
DSM-51,2 defines delusions as fixed beliefs that are not amenable to change in light of conflicting evidence. Of note, the new edition of DSM captured an important nuance, namely that it is the dysfunctional process of belief, rather than its content necessarily being false, which is significant. This definition highlights the central differentiating aspects of delusions, which allow us to identify them in the clinic and distinguish them from other kinds of aberrant beliefs, including non-pathological ones: in particular, the fixedness of the belief (conviction) and cultural non-appropriateness (idiosyncrasy). Another important aspect of delusional belief is its specificity: delusions tend to cluster into certain themes, such as persecutory, grandiose, erotomanic, or somatic. That these themes are constant across cultures suggests an underlying biology. Furthermore, it has long been recognized that there is a difference between somatic delusions—e.g. Capgras, Fregoli, reduplication, or arguably, neglect syndromes, in which the patient is often remarkably disinterested in connecting to other beliefs and experiences—and the themed delusions named previously.
Both hallucinations and delusions are present in many neuropsychiatric illnesses. The classical definition of delusions as false beliefs, and hallucinations as perceptions without corresponding external stimuli, have been criticized as oversimplistic3. Delusions only rarely occur in isolation from other psychotic symptoms4, although this does occur in the so-called delusional disorder2,5. Typically, research on delusions has focused on the wide palette of schizophrenia spectrum disorders, but the basic mechanisms of delusion formation are shared among multiple disease states6. In particular, delusions occur in Alzheimer, Parkinson, and Huntington diseases (HDs), delirium, post-ictal states and bipolar disorder among others, different illnesses presenting with a differential predominance of delusional themes and duration of symptoms, and sometimes correlating with other symptoms such as perceptual disturbances7.
Dysfunction of the dopaminergic system has traditionally been assigned the central role in the pathogenesis of psychotic symptoms8. In health, dopamine (DA) neurotransmission mediates the motivational salience of environmental rewards. In psychosis, salience appears not as a result of exogenous stimuli but rather stems from “an endogenously driven assignment of novelty and salience to stimuli”8,9. Anatomically, aberrant salience is mediated by midbrain dopaminergic projections to mesocorticolimbic areas8,9. It is for this reason that among the illnesses discussed here, dementia in AD is particularly interesting, because it is primarily a cholinergic disturbance10.
Somatic delusions frequently occur in psychotic patients where they accrete more polythematic-like explanations, but they can also occur outside a network of other delusional beliefs, remarkably like the syndromes observed in neurological patients with imageable lesions in their right frontal lobe11. The two-factor hypothesis of delusions12,13,14,15,16 accounts for these differences and puts forward a framework that synthesizes imaging work and recent models of delusional thinking, focusing on right frontal hypofunction and dorsal striatal hyperfunction, and accounting for specific features of delusions.
The effects of antipsychotics on the reasoning bias (“jumping to conclusions”, reduced belief flexibility, an externalized attributional style, and deficits in “theory of mind”) are well known. Antipsychotics improve “theory of mind” and mainly belief flexibility, with little effect on the “jumping to conclusions” trait17. This review will not focus on the therapeutics of delusions which include medications and special types of psychotherapy, for which the reader is referred to an excellent review on the subject18.
The molecular and circuit levels
Functional roles of the cholinergic system in mammalian cognitive processes
Neuronal nicotinic acetylcholine receptors (nAChRs) are a heterogeneous family within the superfamily of pentameric ligand-gated ion channels (pLGIC) that are expressed throughout the brain and involved in a wide range of physiological and physiopathological processes19,20,21,22,23,24. pLGIC include not only the neuronal subtype but also the muscle-type nAChRs, γ-amino butyric acid (GABA)A, glycine, and the 5-HT3 subtype of serotonin receptors.
Although brain nAChRs share a common structural architecture, their cerebral localization, pharmacology, biophysical properties, and developmental occurrence differ. The homomeric (α7 and α9) or heteromeric (α2–α7 combined with β2–β4) nAChR subtypes result from the combination of these subunits into pentameric oligomers of ca. 270,000 Da (see reviews in refs 24,25). Through this combinatorial procedure, Nature has developed abundant diversity to fulfill connectomically and topologically distinct functional requirements. Brain nAChRs have been classically associated with excitatory neurotransmission26 and its modulation (see ref. 27), but these are only fragmentary facets of their functional palette; their cellular distribution along development and adulthood28,29 and subcellular localization (predominantly presynaptic, but also perisynaptic and postsynaptic30; reviewed in ref. 24) adds complexity to this apparently primary role. Thus, nAChRs can modulate the release of other neurotransmitters31,32, or acetylcholine (ACh) itself33,34,35, regulate synapse development36, cell viability37, neurite outgrowth38 and brain energetic homeostasis39, and modulate the establishment of functional networks during neural development40 (see review in ref. 41), apparently affecting the neuronal survival/death homeostasis. Because of their strategic location in areas involved in memory acquisition, maintenance and retrieval, nAChRs in hippocampus and cerebral cortex play major roles in cognition and mnemonic functions42,43 and modulate prefrontal cortex functions related to conscious processing44. All these phenomena are subject to dysfunction under disease conditions (see below). At present, we probably know only a minute fraction of the roles played by native nAChR subtypes in specific functions in the human brain.
The most widely expressed subtypes in brain are the heteromeric α4β2-subtype and the homomeric α7 nAChRs24. These receptors are essentially ionotropic proteins, i.e. ion channels, whose main ability is to rapidly permeate potassium, sodium, and calcium ions in neurons and astrocytes. It has recently been disclosed that α7 nAChRs have an additional property45,46, namely the ability to activate downstream metabolic cascades much like the G-protein-coupled receptors, which constitute a structurally and evolutionarily unrelated superfamily of receptors. This dual functionality only found so far in the α7 nAChR, couples the ionotropic activation elicited by the endogenous agonist ACh to metabotropic neuronal signaling and plasticity phenomena mediated by a G-protein-binding domain located in the intracellular loop of the α7 nAChR. Thus, neuronal α7 nAChRs appear to display both ionotropic and metabotropic properties, whereas α7 nAChR in microglia lack ion channel permeation and exhibit only the metabotropic function, as is the case with other non-neuronal cells, like macrophages, which operate via the JAK2/STAT3 transcription factor pathway, as reviewed elsewhere24.
The α7 nAChR, encoded by the CHRNA7 gene47, has been the prototype of the homomeric (five apparently identical copies, α7 × 5) type of neuronal nAChRs. Another peculiarity of the α7 nAChR is that it possesses five identical ACh recognition sites instead of the canonical two sites of the other nAChRs: heteromeric subtypes carry the two orthosteric agonist-recognition sites at the interfaces between two adjacent non-homologous subunits, one of which is an α-subunit. Gotti and coworkers have characterized a native heteromeric α7β2 nAChR expressed in human basal forebrain and having physiological and pharmacological profiles different from those of the typical homomeric α7 nAChR48 reviewed in refs. 24,49. The latter subtype of neuronal nAChR is highly expressed in brain, particularly in cerebral cortex, subcortical areas, and hippocampus, where it has been associated with mechanisms of neuroprotection and the processes of learning, memory, attention, reward, sensory information processing, and cognition50,51.
Recent experimental work has provided new information on the distribution of nAChRs across the different layers of the cerebral cortex and the different functional roles played by these layer-specific receptors, either located on dendrites of principal neurons or on GABAergic interneurons52. An interesting finding is that the spike timing-dependent synaptic plasticity is oppositely regulated by the activation of nAChRs located in different cortical layers: superficial layer 2/3 (L2/3) pyramidal neurons are inhibited by nAChR activation on interneurons, whereas deep L6 pyramidal neurons are excited by postsynaptic nAChRs. Thus, this stratified nAChR expression allows functional layer-specific control of cortical processing and plasticity by the basal forebrain cholinergic neurons. This system appears to be evolutionarily conserved from mice to humans, the neocortex of the latter maintaining opposite layer-specific cholinergic control of synaptic plasticity. Different sets of cholinergic neurons located in the basal forebrain preferentially target superficial or deep cortical layers of the medial prefrontal cortex (mPFC)52.
Abundant layer-specific neuroanatomical representation of these projections in brain are an important manifestation of phylogenetic evolution; however, it is the diversity and extent of expression of nAChR subtypes at the cellular and subcellular levels, and the multiple combinations of subunits in this pentameric receptor, that play a major role in the physiology and pathology of the nicotinic cholinergic system in brain. The correlation between layer-specific nAChR expression and functional layer-specific control of cortical processing and plasticity by the basal forebrain cholinergic neurons52 is a clear example of the strategy developed by the brain in the course of evolution to maximize structural–functional diversity at the cellular level. The nine α subunits and three β subunits so far identified in the CNS already provide combinatorial potential, which to date, however, appears not to be fully realized in the actual brain. This repertoire of combinatorial possibilities also determines various levels of affinity for the neurotransmitter and cholinomimetic drugs as well as the topographical specialization of nAChRs across the brain architecture, the ion permeability, and various degrees of desensitization resulting from ligand binding to different nAChR subtypes. For instance, the α7 nAChR has an unusually fast on-rate of desensitization, which apparently plays a functional role in the termination of synaptic transmission mediated by this subtype of receptors. The natural ligand, ACh, and the full agonist nicotine trigger sodium flux through the α4β2 nAChR and calcium-mediated currents in the case of the homo-pentameric α7 nAChR, albeit with 100–1000 lower affinity in the latter case53.
Abnormal expression of α4β2 nAChRs has been shown to alter cholinergic neurotransmission in neuropsychiatric disorders, including autism spectrum disorders54,55, nicotine addiction56,57,58,59,60, AD61, and Parkinson disease (PD)56. Alterations of α7 nAChR expression have been associated with various brain disorders, interactions with amyloid-β, and with the pathogenesis of dementia through multiple mechanisms62,63,64,65,66,67,68,69. Expression of the α7 nAChR, which binds nicotine with low affinity, is reduced in the hippocampus of schizophrenic patients70. Early studies found a non-uniform decline in α7 nAChR expression; regional specificity was suggested by the diminution of expression in frontal cortex but not in parietal cortex71.
The experimental evidence accumulated during the last two decades provides solid support to the notion that α7 nAChR is a strong candidate for understanding the pathophysiology of psychosis in both schizophrenia spectrum disorders and AD72,73. Abnormal expression of α7 nAChR has also been associated with the auditory sensory gating deficit characteristically found in patients suffering some forms of schizophrenia spectrum disorders and their relatives. The typical sign is the diminished suppression of an auditory-evoked response (P50) to repeated stimuli. The CHRNA7 gene coding for the α7 nAChR protein has been linked to the P50 deficit74. This finding, in conjunction with evidence of familial transmission of this sensory gating deficit, led to a suggested pathogenic role of the gene coding for the α7 nAChR in schizophrenia spectrum disorders75 (see also refs. 76,77,78,79 for recent reviews of genetic factors in these disorders). Hashimoto80 has prompted the use of auditory sensory gating as a translational biomarker.
Additional associations are found between nicotine addiction and risk of dementia. The comorbidity of nicotine dependence is 2–3-fold higher in psychiatric patients in general, but the association between tobacco smoking and schizophrenia is even stronger, as shown in meta-analysis of worldwide studies: 70% of male schizophrenic patients and 40% female patients were found to be smokers81, a prevalence higher than for any other psychiatric disease. Another linkage between the smoking habit and illnesses is that smoking promotes atherosclerosis and vascular disease increases the risk of dementia, whereas nicotine has been shown to exert neuroprotective effects on dementia via α7 nAChRs in preclinical studies82. Antagonists and agonists of α7 nAChRs have been found to regulate the gating or filtering of auditory information in both humans and animal models83,84,85,86,87.
[H3]-nicotine binding, an in vitro biochemical readout of the high affinity nAChRs, was also found to be lower in patients with schizophrenia spectrum disorders and did not increase in response to tobacco use, as observed in control subjects65. It is hypothesized that patients smoke to alleviate cognitive symptoms57. This is taken as evidence that nicotine enhances cognition88. The hypothesis is still inconclusive; see recent review in ref. 89.
A recent study focuses on the relationship at the gene–gene and gene–environment levels of the interactions between CHRNA7 polymorphism, apolipoprotein E (APOE) ε4 carriers, and smoking on risk of dementia. Late-onset AD (LOAD) patients and 115 vascular dementia patients were compared with healthy individuals. Among APOEε4 non-carriers, CHRNA7 rs7179008 and GT haplotype in CHRNA7 block4 variant carriers exhibited significantly decreased risks. These findings suggest that there are gene associations that up-regulate or down-regulate the risk of vascular and non-vascular forms of dementia90.
Role of muscarinic receptors in psychotic disorders
Although this review addresses primarily the role of nicotinic cholinergic neurotransmission in delusional thinking, we will briefly review the brain muscarinic system and the evidence that points towards its involvement in psychotic states91,92,93,94.
Central muscarinic receptors are G-protein-coupled receptors widely distributed in the mammalian brain. They consist of five subtypes termed M1–M5 and called M1-like and M2-like receptors. M1-like receptors comprise the M1, M3, and M5 receptors. These are located post-synaptically, promoting neurotransmission when activated by ACh. M2-like receptors comprise the M2 and M4 receptors and are located both pre-synaptically and post-synaptically, acting mainly as autoreceptors (for detailed reviews of the central muscarinic system see refs 95,96.
The M1 receptor is the most abundant muscarinic receptor in the cortex, the striatum, and the hippocampus. The M3 receptor shows a distribution which parallels that of the M1 receptor, albeit with a lower density. The nucleus basalis of Meynert and the occipital cortex are rich in M2 receptors. M4 receptors are prominent within the striatum and the caudoputamen. The M5 is the muscarinic receptor with the lowest expression levels; it is found in the substantia nigra pars compacta and the ventral tegmental area97,98,99.
The clinical entity known as anticholinergic delirium observed in patients treated with anticholinergic drugs suggests that muscarinic receptors are implied in these psychotic phenomena100,101,102.
In 1972, Janowsky and coworkers advanced a cholinergic–adrenergic hypothesis of mania and depression103. The authors proposed a balance between cholinergic and adrenergic neurotransmission in those areas of the brain involved in affective regulation. They briefly mention the exacerbation of schizophrenic symptoms in patients exposed to organophosphates104, suggesting a muscarinic cholinergic receptor mechanism after cholinesterase inhibition. Due to increased ACh at the synaptic cleft in multiple brain areas after organophosphate exposure, it is difficult to discriminate between muscarinic and nicotinic receptors in the production of this exacerbated symptomatology.
A more direct involvement of muscarinic receptors in the production of psychosis (in this case negative symptoms of schizophrenia) was advanced by Tandon and Greden105. Positive symptoms of schizophrenia are explained by increased mesocortical and mesolimbic dopamine but a compensatory increase in cholinergic mechanisms is operative in the production of negative symptoms such as alogia, avolition/apathy, anhedonia, and attentional deficits105. This involvement of cholinergic mechanisms and more specifically of muscarinic receptors is supported by a study that shows amelioration by the anticholinergic agent trihexiphenidyl of negative symptoms in schizophrenia patients. Trihexiphenidyl binds to the M1 muscarinic receptor106. This may partially explain the elevation in mood, better socialization, and stimulation effected by trihexyphenidyl in this patient population, often subsequently leading to abuse of this medication107. The effect is also observed in neuroleptic-naïve patients indicating a beneficial effect beyond relief of extrapyramidal side effects105.
The evidence for involvement of muscarinic receptors in schizophrenia spectrum disorders is supported by post-mortem, pharmacological, and imaging studies94. In this respect, an important study by Bakker and collaborators108 was able to show a correlation between a reduced binding potential of M1 receptor in the dorsolateral prefrontal cortex, a key area involved in the production of negative symptoms in schizophrenia spectrum disorders. The study was conducted on medication-naïve psychotic patients and used single-photon emission computed tomography with a specific radioligand that measured binding potential of M1 receptors108. The authors were able to show a correlation between lower binding potential of the M1 receptor and poor outcomes in several neurocognitive tests and negative symptoms severity. However, there was no mention of delusional thinking.
In conclusion: it is clear from the published evidence regarding the involvement of muscarinic receptors in psychosis, specifically in schizophrenia spectrum disorders, that these receptors are related to cognitive deficits in attention, working memory, concentration, and executive functions91,93,108, which in turn translate into negative symptoms, and in neurological soft signs57. Although short-lived delusional thinking is certainly a feature of anticholinergic delirium produced by antimuscarinic drugs101,102, it is not clear how muscarinic receptors participate in the genesis and ulterior maintenance of both well-crystallized and systematized delusional thinking.
Cholinergic pathways in the brain
The cholinergic system consists of subcortical as well as cortical domains, organized into two main pathways: (i) the brainstem and (ii) the magnocellular basal forebrain cholinergic system109.
There are two cholinergic nuclei in the brain, as well as a high concentration of cholinergic interneurons in the basal ganglia. Interactions between cholinergic interneurons and dopaminergic neurons in the basal ganglia are well characterized. The striatum contains extremely high concentrations of ACh relative to other areas in brain, the main locus being in the cholinergic interneurons, specifically in the dorsal striatum (caudate nucleus and putamen), which however constitute only 1–2% of all the striatal cellular content. The mesocorticolimbic system connects the neurons of the ventral tegmental area (VTA) with the nucleus accumbens (NAc, in the ventral striatum) and the prefrontal cortex (PFC). The experience-dependent dopamine neurons in the VTA are key elements in the regulation of the brain reward system, whose dysregulation is associated with depression and mood disorders110,111. Excitatory glutamatergic/cholinergic inputs in dopaminergic neurons, as well as inhibitory GABAergic afferents on these cells, are modulated by nAChRs present in cholinergic, glutamatergic, and GABAergic terminals. A direct activation of excitatory postsynaptic nAChRs results in depolarization and firing of dopaminergic neurons111.
In addition, an indirect mechanism involving α7 nAChRs triggers glutamate release in glutamatergic synapses, which in turn elicits activity of dopaminergic neurons in the VTA112.
ACh’s role in delusions in Alzheimer disease (AD)
Forty-one percent of persons with AD experience hallucinations and delusions early in the course of the disorder113. The occurrence of these symptoms is associated with faster cognitive decline114. Of particular relevance to this review, psychotic thinking consists mainly of persecutory and misidentification delusions115. Cholinergic deficits are implicated in the genesis of cognitive and psychotic symptoms in AD116, and these deficits have been associated with α7 nAChRs in both schizophrenia spectrum disorders and AD72,73,74,75,84,85,117,118. Furthermore, the gene coding for the α7 nAChR (CHRNA7) is known to be associated with symptoms of schizophrenia spectrum disorders in linkage and association studies47,119. More specifically, a strong correlation between delusional thinking in AD and a C/T polymorphism located in intron 3 of CHRNA7 was reported by Carson and coworkers72. Delusional symptoms (mainly of the persecutory and misidentification types) were more pronounced in patients homozygous for the T allele than for the CC or the CT genotypes72.
Cholinergic brain pathways are implicated in cognitive processes57,120. The best example is the cognitive decline seen in patients with AD dementia, which is correlated with early degeneration of ACh-synthesizing neurons in the subcortical nuclei of the human basal forebrain121,122,123. New evidence suggests that the cholinergic system may be implicated not only in cognitive deficits but also in the development of psychotic symptoms in this patient population10,101,124.
A biochemical study examining regions of interest in post-mortem brains of patients with Lewy body dementia found an elevated M1-subtype of muscarinic ACh receptor concentration in the fusiform gyrus of the temporal lobe125; this type of receptors has not been found on dopaminergic neurons in the striatum110,123. In relation to this observation, it is instructive that the relationship between muscarinic tone and psychosis is reversed in schizophrenia spectrum disorders, where decreasing muscarinic tone is associated with increasing psychosis126 and agonism with decreasing psychosis127. Although the exact mechanism of action by which cholinergic deficits produce psychosis in AD is not known, imaging studies show that psychosis is associated with abnormalities in frontal and temporal cortices where cholinergic deficits are more pronounced124. The same brain areas show increased M2-type muscarinic receptor binding128, suggesting participation of these receptors in psychosis. It has been suggested that deficits in pathways involving muscarinic receptors abnormally facilitate dopaminergic systems producing psychosis129. Blockade of cholinergic circuits can produce psychosis, as seen in patients treated with anticholinergic drugs102. Anticholinergic drug-induced delirium, another example of psychosis seen in clinical settings, manifests with frank delusional thinking124. Drugs that enhance cholinergic transmission, such as cholinesterase inhibitors, decrease delusions and hallucinations in AD patients130,131.
ACh in schizophrenia spectrum disorders
The relationship between nicotinic signaling and schizophrenia is complex, given the number of subunit combinations of this neurotransmitter receptor possibly involved and their distribution within the central nervous system. However, although the involvement of nicotinic signaling in the general cognitive decline in schizophrenia spectrum disorders is clear57,120,132, as is its potential to modestly improve cognition, this alone is insufficient to suggest participation of the nicotinic type of neurotransmission in delusions, as there is no consensus on the relationship between cognitive deficits of schizophrenia and the genesis and maintenance of delusional thinking133,134. It is also clear that a4β2 receptors are specifically upregulated in the striatum in smokers in general but not at all or at least not to the same degree in smokers with schizophrenia spectrum disorders135,136.
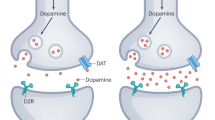
In connection with the dopaminergic hypothesis of psychosis, research on addiction has shown that nicotine drives a much greater increase in phasic signaling by DA in the NAc than in the dorsal striatum which contains the caudate nucleus and putamen137; furthermore, it has been shown that there is a reduced density of cholinergic interneurons in the ventral striatum in schizophrenia spectrum disorders138,139. The NAc expresses α6 subunit-containing nAChRs, rather than the α4 subunit-containing receptors that constitute the majority of dorsal striatum nAChRs140. Notably, a consistent finding has been that of nicotine serving the role of reinforcing reward rather than affecting the dorsal striatum where the putative central cognitive pattern-generating striosomes reside (see Fig. 1). This is consistent with nicotine being addictive but having no effect on positive symptoms and with its documented effect on mesolimbic circuitry and brain regions associated with reinforced behavior, rather than feedback-insensitive behaviors like those produced by central pattern generators. Mesolimbic modulation is also consistent with improvement in the negative symptoms sometimes seen with nicotinic agonism141 but not in positive symptoms. α7 nAChR agonists have also been shown to improve both negative symptoms and cognition in schizophrenia spectrum disorders142. Notably, it has been shown that cholinergic interneurons in the dorsal striatum are responsible for this modulation143. Furthermore, the linkage of α7 nAChRs (widely distributed in cortex) with schizophrenia spectrum disorders has been established for some time by GWAS studies88.

Striosomes are the site of integration of input from the substantia nigra pars compacta (SNc), thalamus, and cortex, and are prime candidates for the central cognitive pattern generators whose dysfunction may underlie delusions. The matrix, composed primarily of cholinergic interneurons, surrounds the striosomes and modulates input. Output from the striatum is through medium spiny neurons which target the internal globus pallidus (GPi) or the external segment of the globus pallidus (GPe) and eventually, the thalamus146,147.
The model proposing dopaminergic projections from the VTA to the ventral striatum (the mesolimbic circuits) is no longer well-supported, as shown by the meta-analysis of positron emission tomography (PET) data and a review of all available imaging and anatomic data144,145. A more likely picture is the central involvement of dysfunction of the nigrostriatal circuits which make synaptic contacts with striosomes in the dorsal striatum. Striosomes are histochemically recognizable striatal compartments within the striatum146,147,148 and will be a key region to further discuss in terms of their putative involvement in delusional thinking (see Figs 1 and 2).

Dopaminergic inputs are received from the SNc and glutamatergic inputs from the cortex and thalamus. The cholinergic system has a key role in the integration of these signals, which are modulated by cholinergic interneurons in the matrix surrounding the striosomes, and output is mediated by medium spiny neurons, which are GABAergic neurons forming the direct and indirect pathways (thick vertical arrows) depicted here and in Fig. 1. See also refs 146,147,230.
Cholinergic neuronal cells represent only 2% of cells in the striatum but appear to establish synapses on a large fraction of striatal cells149. There is anatomical and pharmacologic evidence for this role. By identifying cells in the striatum with choline acetyltransferase (ChAT) immunoreactivity, it was shown that they are most frequently found in the periphery of striosomes where they are ideally positioned to modulate nigrostriatal dopaminergic inputs; there are far fewer in the putamen150. Muscarinic M4-agonism alleviates the amphetamine model of psychoses in rodents, with the dorsal striatum as a preferential target127. It has been shown that medication-naive patients with schizophrenia spectrum disorders have reduced levels of muscarinic receptors, particularly in the striatum, as to be expected in individuals having difficulty adapting to uncertainty and rapidly updating beliefs126.
Striosomes are the most likely central pattern generators, e.g. responsible for species-specific stereotypies. If some delusions are indeed cognitive stereotypies arising from hyperfunction of striosomes functioning as cognitive central pattern generators, we can make strong inferences and predictions about the behavior of delusions under various experimental manipulations (see Fig. 3). Stereotypies are well known to be intensified or prolonged by increasing dopaminergic input to, or tone in, the striatum, for example in amphetamine paradigms, both experimentally and in stimulant abuse in humans. Increasing cholinergic transmission in the dorsal striatum stops stereotypies more quickly; decreasing cholinergic transmission by blocking postsynaptic receptors with muscarinic antagonists increases the length of stereotypies151 and striatal cholinergic interneurons modulate hyperdopaminergic dyskinesias152.

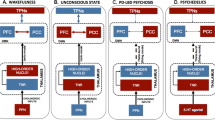
While it is not a comprehensive diagram of all receptors or all interactions between these neurons, the diagram is intended to highlight the inputs from the cholinergic interneurons, as well as the cortex, thalamus, and substantia nigra pars compacta (SNc), all converging on the medium spiny neuron. Importantly, here is where ACh exerts its effect on prediction error, modulating prediction error by boosting bottom-up signals.
This picture is complicated by the differential function of muscarinic and nicotinic receptors. In the striata of patients with schizophrenia spectrum disorders there are fewer M1-type muscarinic receptors153. M1 receptors are not located on cholinergic interneurons and they do not serve an autoreceptor function154. However, there are presynaptic non-M1 receptors on cholinergic interneurons154. There is also greater nicotine binding in schizophrenic patients, presumably on the postsynaptic target neurons155. One would therefore expect that increasing muscarinic or decreasing nicotinic neurotransmission would improve patients suffering from delusions. The striatum is primarily composed of two classes of spiny projection neurons (SPNs): the striatonigral and striatopallidal SPNs, which express D1R and D2R, respectively. Within striosomes there are more D1R-expressing than D2R-expressing SPNs, whereas in the matrix there are more D2R-expressing than D1R-expressing SPNs (Figs 2 and 3).
ACh in delusional illness: PD
Psychosis is a non-motor and late complication of PD. It predicts Lewy body pathology in the brain cortex156 and shows a variable response to antipsychotic agents. In fact, psychosis in PD presents with marked heterogeneity suggesting the involvement of multiple neurotransmitter systems in its genesis. Recently, Factor and coworkers were able to delineate multiple subtypes of PD psychosis157. Delusions are present in about 6–16% of patients, with Othello syndrome, grandiose, religious and guilt types being the most frequent forms of delusion157.
The exact mechanism of delusional symptoms in PD patients is still obscure and is not explained by current ideas on the development of psychosis in these patients158. However, the role of the cholinergic system in PD is made clear by the finding of a strong, statistically significant correlation between hallucinations (but not delusions) and freezing of gait159, both coexisting with cognitive decline. Cholinergic mechanisms are involved in the development of hallucinations160 but, interestingly, delusions are not associated with cognitive decline161 and some patients present with pure delusional syndromes in the absence of perceptual disturbances162,163. The development of delusions does not appear to have a cholinergic basis, is related to dopaminergic medications, and is reversible upon discontinuation of the offending drug164.
ACh in other neuropsychiatric illnesses
It should be noted that although the pathophysiology of delirium is thought to rely on a disturbance in cholinergic neurotransmission165, given the acuity and rapid course of the illness and medical fragility of those suffering from it, there is a dearth of imaging studies directly examining this question and certainly in looking at the specific pathogenesis of delusions in this condition166,167. It is intriguing that there is a case report of a patient with delirium secondary to a urinary tract infection developing a Capgras delusion, presumably as part of delirium, which resolved in days with the cure of the organic illness168.
The neuronal cell and circuitry levels
Huntington disease
The occurrence of neuropsychiatric symptoms in HD, including positive symptoms of psychosis, is common169 and generally under-appreciated. While almost all HD patients develop neuropsychiatric symptoms during their illness, a recent survey found that psychosis is the presenting feature in 11% of cases and that delusions occurred in 11.5% of patients170. Obsessive compulsive symptoms are more common than delusions171, consistent with the theory of striosomal hyperfunction as underlying both cognitive stereotypies (obsessions, delusions) and motor behaviors, as in obsessive compulsive and tic disorders. It is also curious that, similar to patients with psychosis, HD patients often have poor insight into their deficits, both cognitive and motor172.
The etiogenesis of HD is well characterized173, with the expansion of a trinucleotide repeat in the Huntington gene, causing a toxic gene product to accumulate in and cause selective death of the GABAergic medium spiny neurons, making psychotic symptoms difficult to explain in terms of the dopaminergic model of psychosis. These neurons constitute the majority of cells in the caudate174. As a result, as the disease progresses the classic finding is radiologically visible atrophy of the caudate heads, although post-mortem studies typically show degeneration in many brain regions175,176,177,178. While in terms of cell count, cholinergic interneurons are spared, ChAT levels drop, suggesting dysfunction of these cells179. Consistent with the current model of the computational function of striatal cholinergic interneurons, with decreasing activity of these cells we should expect an intensification of stereotypies and rigid beliefs. Therefore, a test of the cholinergic neurotransmission aspect of the striosomal hypothesis of delusions would be to investigate the association between the extent or distribution of ChAT loss and the onset or intensity of psychiatric symptoms including delusions; to the best of our knowledge no such data is available as yet. Similarly, delirium and many dementing illnesses are canonically considered to be disruptions in ACh signaling. To our knowledge, there has been no attempt to link ACh signaling directly in the basal nuclei, and the caudate in particular, to the severity of the pathology in these illnesses, including delusions. Given the increasing importance of this nucleus in neuropsychiatric illness and the argument here for its role in delusion, this aspect should be further explored.
ACh and prediction error
A great deal of what we know about cognitive approaches to understanding delusions has been pioneered by the work of Corlett12,13,14,15,16. Brains are inference engines which construct hypotheses to predict or interpret stimuli (“top-down” processing), and when there is a discrepancy between prediction and perception, generate an error message (a prediction error signal, “bottom-up” processing)180,181, see review in ref. 182. This Bayesian (probabilistic) inference mechanism is understood to be defective in various forms of psychoses, particularly in schizophrenia spectrum disorders, either because of abnormal signaling of the prediction errors, abnormal sensory inputs, or both, ultimately resulting in dysfunctional predictive coding. Applying this concept to the empirical dysfunction observed in delusional illnesses may show where this computation is instantiated and ultimately suggest corrective measures that could be used in therapeutic approaches.
Thus far, research on the prediction error signal has largely focused on glutamatergic or dopaminergic neurotransmission. Predictive coding has been associated with gain of control of postsynaptic glutamatergic excitatory transmission mediated by NMDA receptors and dopaminergic neuromodulation180. However, as schematically depicted in Fig. 3, ACh has an important role in modulating this signal; the behavior of VTA circuitry is explained by a computational model showing that the endogenous neurotransmitter ACh and the exogenous full nicotinic agonist nicotine both effectively desensitize nAChRs and cause increased reward signals, i.e. decreasing ability to predict reward183. These effects are mediated by α4β2 nAChRs on GABAergic neurons acting on dopaminergic neurons—also containing α4β2 nAChRs on their surface—in the VTA (reviewed in ref. 183). This has been demonstrated in an experimental paradigm using galantamine application in combination with EEG measurements, by showing that ACh boosts bottom-up prediction errors, which are preferentially observed under conditions of increased uncertainty184,185. In this way the brain’s purported hierarchical Bayesian prediction function can be tuned to increase input from top-down or bottom-up data, for optimal predictions under varying conditions186. It is intriguing that one of the hallmarks of delusional ideation is pathologic conviction (certainty when it is not justified), especially as it relates to the striosomal hypothesis of delusions. Yamanaka and coworkers argue that thalamostriatal neurons project to striatal cholinergic interneurons, and that this system generates an “associability” signal, the certainty of which is modulated by cholinergic neurotransmission187. It is known that dopamine release from these thalamocortical projections is tonic and suppresses firing of striatal cholinergic interneurons, but the significance of this in the context of illness is unclear188.
It is not clear whether ACh can be accurately measured by magnetic resonance spectroscopy (MRS) in awake subjects. MRS allows the measurement of molecules in the brain by detecting the specific magnetic fields of protons as they are shifted by moieties in those molecules. While two neurotransmitters (glutamate and GABA) can be accurately and reliably measured, neurochemical and technical difficulties have so far made this challenging for ACh. First, it is the choline moiety (not the full ACh molecule) that is detected, and a substantial fraction (likely the majority) of this signal can be assumed to come from free choline being used for cell membrane biosynthesis. Second, there is less ACh present than the two other neurotransmitters mentioned, and this worsens the signal to noise ratio. Nonetheless, in rat, MRS-measured choline was shown to correlate with directly (chemically) measured ACh189. However, functional changes may not be measurable, as task-driven changes in MRS-measured choline were similar to the presumed unrelated metabolite N-acetyl-aspartate, suggesting that dynamic MRS measurements may still not be sufficiently sensitive and/or reliable190.
Linking up with the framework of Bayesian reasoning and prediction error, it has been argued that ACh modulates prediction error signals and tunes the brain’s updating in response to varying degrees of uncertainty. That is, in predictable environments, ACh enhances bottom-up signaling (to enhance the salience of sense data) and in unpredictable environments, it dampens it. This model is experimentally supported by increased sensory cortex response in an oddball paradigm with human subjects taking galantamine184. Galantamine also increases the rate of updating in a spatial beliefs model (using an eye-tracking paradigm)185. Consequently, a hypocholinergic state would be expected to slow belief updating (as in delusional belief) and an increase in cholinergic tone would improve it, as shown with galantamine191. Acetylcholinesterase inhibitors (which increase ACh neurotransmission) have been used off-label to improve cognition in schizophrenia192,193,194,195. The obvious inference is that the decreased cholinergic neurotransmission, consistent with what is seen in α7 nAChR loss-of-function mutations in animal models, would result in decreased ability to adapt to environments based on predictability and consistent low weight placed on unexpected data in predictable environments, relative to top-down beliefs. Cognitively, this could be envisioned as someone who is convinced of the predictability of the world despite sense data inconsistent with this belief.
An additional role of ACh is worth mentioning. In a seminal paper, Moran and coworkers showed that ACh is a neuromodulatory transmitter involved in perception and learning under uncertainty184. Using computational simulations and electroencephalography the authors suggested that ACh enhances the precision of bottom-up synaptic transmission, optimizing the gain of pyramidal cells184,185.
A unifying hypothesis: delusions are in part caused by hyperdopaminergia and overactive striosomes in the dorsal striatum, modulated by cholinergic interneurons
One shortcoming of delusion theories thus far is the minimal effort taken to explain the specificity of polythematic content, with delusion being clustered into predictable themes across cultures. The dopamine theory of psychosis is well-established; dopamine blockade improves positive symptoms and dopamine agonists induce or worsen positive symptoms. What remains under-appreciated is the anatomy of dopamine modulation in psychosis, with recent evidence pointing toward the nigrostriatal dopaminergic pathway and focusing on the dorsal striatum as relevant in the production of psychotic symptoms145. Specific models of delusional belief show that changes in activity in the dorsal striatum as observed with fMRI correlate with the amount of delusional ideation in illness196.
One argument for the source of delusional themes advances striosomes as the explanation for the content of delusions148, conserved as they are cross-culturally, strongly suggesting a biological basis. As illustrated in Figs. 1–3, striosomes are clusters of specialized neurons within the caudate which receive dopaminergic projections from the substantia nigra pars compacta148. Striosomes are known to be responsible for specific motor programs (stereotypies) which have clear evolutionary importance and are not subject to modification and are highly reward-insensitive. For example, individual striosomes in rodents have been isolated and shown to be responsible for specific stereotypies, e.g. parts of the grooming chain197. The basal ganglia are evolutionarily older than the cortex and off-loading repetitive, evolutionarily important actions makes economic sense for the brain. Dopamine agonism has been shown to cause growth in the striosomes, the degree of which in turn is correlated with the increase in stereotypy frequency198,199.
While thus far striosomes have only been shown to be responsible for motor programs in non-human animals, it is speculated that in humans they may also be responsible not only for stereotypies, but also for cognitions which are of similar evolutionary importance and reward-insensitive. The fit with delusional themes is immediately apparent—for instance, identification of conspiring competitors, possible mates, or cheating on one’s own mate. Furthermore, delusions are responsive to dopamine blockade or agonism, and in other illnesses like OCD or Tourette syndrome, there is a clear crossover between repetitive motor and cognitive patterns200. Some of the themes of OCD overlap with delusions and indeed sometimes there is damaged, but not absent, insight, making diagnosis difficult and resulting in discussion at one time about adding a schizo-obsessive diagnostic category to DSM201,202. Tourette is a related illness with a specific reward-insensitive behavior that often responds to antipsychotics. In cases where it does not, the nicotinic antagonist mecamylamine has been found to sometimes be effective203. The argument is strengthened by our increasingly detailed knowledge of the complex architecture of the dorsal striatum, with considerable evidence that matrix and striosome respond differently to the same inputs147,204. There are differences depending on nicotinic receptor subtype within the striosomes, and differential activation may be what drives fixed motor pattern (stereotypies)205. The role in aberrant movement of dopaminergic hyperstimulation of the dorsal striatum and its modulation by cholinergic inputs has been well-established152. In fact in mice with amphetamine-induced stereotypies and poor response to environmental cues, direct electrophysiology shows that cholinergic modulation of striosomes is disrupted, again suggesting that ACh in the dorsal striatum serves to increase sensitivity to bottom-up information and thus predispose to belief updating. This has an obvious parallel in amphetamine-induced psychosis in humans, as humans develop tics and stereotypical behaviors in response to intoxication with dopamine agonists206. If as has previously been argued207 we can extend the animal models of striosome-originated stereotypies to delusions, taking stereotypies as rigid behaviors, a kind of “motor delusion” (or considering delusions as “cognitive stereotypies”), then this would explain the neuroanatomy underlying delusions in psychosis and dopamine agonist intoxication. The striosomal model therefore argues that the specific themes of delusions (specificity), and their insensitivity to new information (conviction) is the result of hyperdopaminergic, over-driven striosomes. It should also be noted that computational studies of cognition have noted attractor basin-like dynamics demonstrated by delusion formation208, and striosomes are an excellent candidate for the physical instantiation of this phenomenon.
In terms of neurotransmission, it is suggestive that the highest concentration of ACh in the brain is in fact in the dorsal striatum209, and given the role of this neurotransmitter in modulating striatal prediction error, one would expect cholinergic antagonism to worsen delusions, which in fact is what is observed, particularly in delirium and AD dementia, as well as in some cases of Huntington and iatrogenic psychoses. Thus, the striosomal theory of delusional content accounts both for the dopamine theory of psychosis, and deviations from the model where the disturbance is cholinergic.
It should be noted that the dorsal striatum accounts for only part of the hypothesis; namely, the specificity of polythematic delusions. The poor insight present in delusions, as well as somatic and misidentification delusions, very likely result from functional lesions of the right frontal lobe. Indeed, that somatic or misidentification delusions (e.g. Capgras syndrome) result from lesions of the right frontal lobe is one of the best supported observations about the anatomy of delusions so far210,211. Given these putative roles, it may be the case that a right frontal lesion without striosomal hyperfunction results in somatic delusions, whereas striosomal hyperactivity in the dorsal striatum occurring without right frontal deficits would induce thematically specific cognitive or motor behaviors, with preserved insight, conditions that are worsened by dopamine agonists and improved by dopamine antagonists. This is a description of OCD and Tourette syndrome, which we know involve disruptions to striatal signaling212 and often respond clinically to dopamine blockade.
It is important to note that the cognitive approaches for understanding delusional thinking using a two-factor hypothesis, mainly proposed by Corlett12,13,14,15,16, have been recently revisited by this author213.
New directions for investigating and treating delusions
Over the past two decades there has been a focus on developing cholinergic (especially nicotinic) modulators for schizophrenia spectrum disorders and AD that have yielded inconsistent results214, as well as case reports with acetylcholinesterase inhibitors193,215. It may be that the best role for such molecules is as an adjunct, given the cholinergic system’s role in modulating uncertainty and updating, while dopamine is more central to the etiology of psychosis. Recent pharmacological evidence has provided new insights into our understanding of cholinergic signaling in brain and have put the α4β2 and the α7 nAChRs in a center-stage position as possible targets for therapeutic intervention in neuropsychiatric diseases with cognitive deficits including autism spectrum disorders, nicotine addiction, schizophrenia spectrum disorders88, AD, and PD56,216,217,218. For instance, rivastigmine, a drug that increases cholinergic tone by inhibiting the enzyme cholinesterase, is effective for dementia, whereas the use of Donepezil is still in the realm of investigation218. Activation of the α7 nAChRs by selective ligands enhances cognition in schizophrenic and AD patients74,84,85,219.
Another set of compounds gaining momentum is the wide spectrum of allosteric modulators, new leads as therapeutic agents for the treatment of neuropsychiatric diseases with cognitive deficits220. These ligands exert their action in the presence of the endogenous agonist, and by definition do not target the canonical (“orthosteric”) nAChR agonist or competitive antagonist recognition sites on the receptor molecule, but bind to distant, non-orthosteric, allosteric sites which nonetheless affect the response of the orthosteric site. There are essentially two types of allosteric modulators, most widely studied for the α7 nAChR, which either potentiate (positive allosteric modulators, “PAMs”) or inhibit (negative allosteric modulators, or “NAMs”) the response of agonist binding to the orthosteric site221,222,223. A third type of compounds corresponds to the “silent” allosteric modulators (“SAMs”), which inhibit the PAM responses without affecting the orthosteric responses. Some authors include a fourth category, namely the allosteric agonists, but clearly these should not be classified as modulators: they are agonists which bind not to the orthosteric but to the allosteric site. Presumably, endogenous allosteric modulators occur in brain. In animal studies, PAMs acting on α7 nAChRs, like PNU-120596 and galantamine, have been shown to enhance performance in various cognitive and recognition memory tasks224. The prevalent effect of PAM compounds is to increase the probability of the nAChR channel open state, thereby increasing its lifetime and the number of open channel events225. Furthermore, the PAM compounds CCMI, PNU120596, and A582941 reverse the sensorimotor gating impairment evoked by MK-801 based on the pre-pulse inhibition of the startle response, thus showing a beneficial effect on sensorimotor gating and some aspects of cognition226.
There is also a great wealth of data on clinical and preclinical observations where muscarinic receptors are targeted. The most conspicuous of these muscarinic agents is xanomeline227 and orthosteric muscarinic ACh receptor agonist which binds to M1 and M4 receptors. It has proven efficacy in reducing psychotic symptoms in schizophrenia228 and AD229, including an effect on delusional thinking. Recently, xamomeline has been used in conjunction with tropsium, a peripheral muscarinic antagonist in hospitalized patients with schizophrenia that experience an exacerbation of symptoms. This was a randomized, double-blinded, placebo-controlled Phase-2 trial (clinicaltrials.gov, NCT03697252).
References
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders. in Diagnostic Statistical Manual of Mental Disorders, 5th edn (Arlington, VA: American Psychiatric Association, 2013).
American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5 5th edn (American Psychiatric Association, 2013).
Humpston, C. S. et al. From computation to the first-person: auditory-verbal hallucinations and delusions of thought interference in schizophrenia-spectrum psychoses. Schizophrenia Bull. 45, S56–S66 (2019).
Barch, D. M. et al. Logic and justification for dimensional assessment of symptoms and related clinical phenomena in psychosis: relevance to DSM-5. Schizophrenia Res. 150, 15–20 (2013).
Kendler, K. S. Demography of paranoid psychosis (delusional disorder): a review and comparison with schizophrenia and affective illness. Arch. Gen. Psychiatry 39, 890–902 (1982).
Bebbington, P. & Freeman, D. Transdiagnostic extension of delusions: schizophrenia and beyond. Schizophrenia Bull. 43, 273–282 (2017).
Kulick, C. V., Montgomery, K. M. & Nirenberg, M. J. Comprehensive identification of delusions and olfactory, tactile, gustatory, and minor hallucinations in Parkinson’s disease psychosis. Parkinsonism Relat. Disord. 54, 40–45 (2018).
Kapur, S. How antipsychotics become anti-“psychotic”-from dopamine to salience to psychosis. Trends Pharmacol. Sci. 25, 402–406 (2004).
Kapur, S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 160, 13–23 (2003).
Cummings, J. L. & Back, C. The cholinergic hypothesis of neuropsychiatric symptoms in Alzheimer’s disease. Am. J. Geriatr. Psychiatry 6, S64–S78 (1998).
Devinsky, O. Delusional misidentifications and duplications: right brain lesions, left brain delusions. Neurology 72, 80–87 (2009).
Corlett, P. R. & Fletcher, P. C. Delusions and prediction error: clarifying the roles of behavioural and brain responses. Cogn. Neuropsychiatry 20, 95–105 (2015).
Corlett, P. R., Frith, C. D. & Fletcher, P. C. From drugs to deprivation: a Bayesian framework for understanding models of psychosis. Psychopharmacology 206, 515–530 (2009).
Corlett, P. R., Honey, G. D. & Fletcher, P. C. From prediction error to psychosis: ketamine as a pharmacological model of delusions. J. Psychopharmacol. 21, 238–252 (2007).
Corlett, P. R. et al. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain 130, 2387–2400 (2007).
Corlett, P. R., Taylor, J. R., Wang, X. J., Fletcher, P. C. & Krystal, J. H. Toward a neurobiology of delusions. Prog. Neurobiol. 92, 345–369 (2010).
So, S. H., Garety, P. A., Peters, E. R. & Kapur, S. Do antipsychotics improve reasoning biases? A review. Psychosom. Med. 72, 681–693 (2010).
Skelton, M., Khokhar, W. A. & Thacker, S. P. Treatments for delusional disorder. Cochrane Database Syst. Rev. Cd009785, https://doi.org/10.1002/14651858.CD009785.pub2 (2015).
Gotti, C. C. et al. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharm. 78, 703–711 (2009).
Changeux, J.-P. et al. Nicotinic receptors and brain plasticity. Cold Spring Harb. Symp. Quant. Biol. 61, 343–362 (1996).
Changeux, J.-P. et al. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res. Rev. 26, 198–216 (1998).
Gotti, C., Riganti, L., Vailati, S. & Clementi, F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr. Pharm. Des. 12, 407–428 (2006).
Taly, A., Corringer, P. J., Guedin, D., Lestage, P. & Changeux, J. P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 8, 733–750 (2009).
Zoli, M., Pucci, S., Vilella, A. & Gotti, C. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 16, 338–349 (2018).
Changeux, J. P. The nicotinic acetylcholine receptor: a typical ‘allosteric machine’. Philos. Trans. R. Soc. Lond. B 373, https://doi.org/10.1098/rstb.2017.0174 (2018).
Alkondon, M., Pereira, E. F. & Albuquerque, E. X. Alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 810, 257–263 (1998).
Yan, Y. et al. Nicotinic cholinergic receptors in VTA glutamate neurons modulate excitatory transmission. Cell Rep. 23, 2236–2244 (2018).
Hellstrom-Lindahl, E., Gorbounova, O., Seiger, A., Mousavi, M. & Nordberg, A. Regional distribution of nicotinic receptors during prenatal development of human brain and spinal cord. Brain Res. Dev. Brain Res. 108, 147–160 (1998).
Murakami, K., Ishikawa, Y. & Sato, F. Localization of alpha7 nicotinic acetylcholine receptor immunoreactivity on GABAergic interneurons in layers I–III of the rat retrosplenial granular cortex. Neuroscience 252, 443–459 (2013).
Caruncho, H. J., Guidotti, A., Lindstrom, J., Costa, E. & Pesold, C. Subcellular localization of the alpha 7 nicotinic receptor in rat cerebellar granule cell layer. Neuroreport 8, 1431–1433 (1997).
el-Bizri, H. & Clarke, P. B. Blockade of nicotinic receptor-mediated release of dopamine from striatal synaptosomes by chlorisondamine administered in vivo. Br. J. Pharm. 111, 414–418 (1994).
Lena, C., Changeux, J. P. & Mulle, C. Evidence for “preterminal” nicotinic receptors on GABAergic axons in the rat interpeduncular nucleus. J. Neurosci. 13, 2680–2688 (1993).
Bali, Z. K., Nagy, L. V. & Hernadi, I. Alpha7 nicotinic acetylcholine receptors play a predominant role in the cholinergic potentiation of N-methyl-d-aspartate evoked firing responses of hippocampal CA1 pyramidal cells. Front. Cell. Neurosci. 11, 271 (2017).
el-Bizri, H. & Clarke, P. B. Regulation of nicotinic receptors in rat brain following quasi-irreversible nicotinic blockade by chlorisondamine and chronic treatment with nicotine. Br. J. Pharm. 113, 917–925 (1994).
Koukouli, F. & Maskos, U. The multiple roles of the alpha7 nicotinic acetylcholine receptor in modulating glutamatergic systems in the normal and diseased nervous system. Biochemical Pharmacol. 97, 378–387 (2015).
Rosenthal, Justin S., Yin, J., Long, C., Spillman, E., Sheng, C. & Yuan, Q. View ORCID profile. Temporal regulation of nicotinic acetylcholine receptor subunits supports central cholinergic synapse development. B-ENT Preprint at https://doi.org/10.1101/790659 (2019).
Voytenko, L. P. et al. Hippocampal GABAergic interneurons coexpressing alpha7-nicotinic receptors and connexin-36 are able to improve neuronal viability under oxygen-glucose deprivation. Brain Res. 1616, 134–145 (2015).
De Jaco, A., Bernardini, L., Rosati, J. & Tata, A. M. Alpha-7 nicotinic receptors in nervous system disorders: from function to therapeutic. Perspect. Cent. Nerv. Syst. Agents Med. Chem. 17, 100–108 (2017).
Stojakovic, A., Espinosa, E. P., Farhad, O. T. & Lutfy, K. Effects of nicotine on homeostatic and hedonic components of food intake. J. Endocrinol. 235, R13–R31 (2017).
Colangelo, C., Shichkova, P., Keller, D., Markram, H. & Ramaswamy, S. Cellular, synaptic and network effects of acetylcholine in the neocortex. Front. Neural Circuits 13, 24 (2019).
Gandelman, J. A., Newhouse, P. & Taylor, W. D. Nicotine and networks: potential for enhancement of mood and cognition in late-life depression. Neurosci. Biobehav. Rev. 84, 289–298 (2018).
Lykhmus, O., Kalashnyk, O., Uspenska, K. & Skok, M. Positive allosteric modulation of Alpha7 nicotinic acetylcholine receptors transiently improves memory but aggravates inflammation in LPS-treated mice. Front. Aging Neurosci. 11, 359 (2019).
Sabec, M. H., Wonnacott, S., Warburton, E. C. & Bashir, Z. I. Nicotinic acetylcholine receptors control encoding and retrieval of associative recognition memory through plasticity in the medial prefrontal cortex. Cell Rep. 22, 3409–3415 (2018).
Koukouli, F., Rooy, M., Changeux, J. P. & Maskos, U. Nicotinic receptors in mouse prefrontal cortex modulate ultraslow fluctuations related to conscious processing. Proc. Natl Acad. Sci. USA 113, 14823–14828 (2016).
King, J. R. & Kabbani, N. Alpha 7 nicotinic receptor coupling to heterotrimeric G proteins modulates RhoA activation, cytoskeletal motility, and structural growth. J. Neurochem. 138, 532–545 (2016).
King, J. R., Nordman, J. C., Bridges, S. P., Lin, M. K. & Kabbani, N. Identification and characterization of a G protein-binding cluster in alpha7 nicotinic acetylcholine receptors. J. Biol. Chem. 290, 20060–20070 (2015).
Sinkus, M. L. et al. The human CHRNA7 and CHRFAM7A genes: a review of the genetics, regulation, and function. Neuropharmacology 96, 274–288 (2015).
Zoli, M., Pistillo, F. & Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96, 302–311 (2015).
Wu, J. et al. Heteromeric alpha7beta2 nicotinic acetylcholine receptors in the brain. Trends Pharmacol. Sci. 37, 562–574 (2016).
Nemecz, A., Prevost, M. S., Menny, A. & Corringer, P. J. Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron 90, 452–470 (2016).
Nees, F. The nicotinic cholinergic system function in the human brain. Neuropharmacology 96, 289–301 (2015).
Verhoog, M. B. et al. Layer-specific cholinergic control of human and mouse cortical synaptic plasticity. Nat. Commun. 7, https://doi.org/10.1038/ncomms12826 (2016).
Le Houezec, J. Nicotine: abused substance and therapeutic agent. J. Psychiatry Neurosci. 23, 95–108 (1998).
Deutsch, S. I., Urbano, M. R., Neumann, S. A., Burket, J. A. & Katz, E. Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin. Neuropharmacol. 33, 114–120 (2010).
Wang, L. et al. Modulation of social deficits and repetitive behaviors in a mouse model of autism: the role of the nicotinic cholinergic system. Psychopharmacology 232, 4303–4316 (2015).
Perez Lloret, S. & Barrantes, F. J. Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. npj Parkinson's Dis. 2, 16001 (2016).
Ochoa, E. L. & Lasalde-Dominicci, J. Cognitive deficits in schizophrenia: focus on neuronal nicotinic acetylcholine receptors and smoking. Cell. Mol. Neurobiol. 27, 609–639 (2007).
Widstrom, R., Aguilar, J. S. & Ochoa, E. L. M. Nicotine up-regulation of nicotine binding sites in NGF-differentiated PC12 cells: correlation with nicotine-induced Ach release. Soc. Neurosci. Abstr. 21, 1330 (1995).
Vibat, C. R., Lasalde, J. A., McNamee, M. G. & Ochoa, E. L. M. Differential desensitization properties of rat neuronal nicotinic acetylcholine receptor subunit combinations expressed in Xenopus laevis oocytes. Cell. Mol. Neurobiol. 15, 411–425 (1995).
Ochoa, E. L. Nicotine-related brain disorders: the neurobiological basis of nicotine dependence. Cell. Mol. Neurobiol. 14, 195–225 (1994).
Lombardo, S. & Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 96, 255–262 (2015).
Nordberg, A., Alafuzoff, I. & Winblad, B. Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J. Neurosci. Res. 31, 103–111 (1992).
Nordberg, A. Human nicotinic receptors—their role in aging and dementia. Neurochem. Int. 25, 93–97 (1994).
Leonard, S. et al. Nicotinic receptor function in schizophrenia. Schizophr. Bull. 22, 431–445 (1996).
Leonard, S. et al. Nicotinic receptors, smoking and schizophrenia. Restor. Neurol. Neurosci. 12, 195–201 (1998).
Mexal, S. et al. Differential regulation of alpha7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J. Mol. Neurosci. 40, 185–195 (2010).
Lewis, A. S. & Picciotto, M. R. High-affinity nicotinic acetylcholine receptor expression and trafficking abnormalities in psychiatric illness. Psychopharmacology 229, 477–485 (2013).
Weiland, S., Bertrand, D. & Leonard, S. Neuronal nicotinic acetylcholine receptors: from the gene to the disease. Behav. Brain Res. 113, 43–56 (2000).
Palma, E., Conti, L., Roseti, C. & Limatola, C. Novel approaches to study the involvement of alpha7-nAChR in human diseases. Curr. Drug Targets 13, 579–586 (2012).
Dineley, K. T., Pandya, A. A. & Yakel, J. L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 36, 96–108 (2015).
Guan, Z. Z., Zhang, X., Blennow, K. & Nordberg, A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport 10, 1779–1782 (1999).
Carson, R. et al. Genetic variation in the alpha 7 nicotinic acetylcholine receptor is associated with delusional symptoms in Alzheimer’s disease. Neuromol. Med. 10, 377–384 (2008).
Tregellas, J. R. et al. Effects of an alpha 7-nicotinic agonist on default network activity in schizophrenia. Biol. Psychiatry 69, 7–11 (2011).
Olincy, A. et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch. Gen. Psychiatry 63, 630–638 (2006).
Adler, L. E. et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr. Bull. 24, 189–202 (1998).
Dennison, C. A., Legge, S. E., Pardinas, A. F. & Walters, J. T. R. Genome-wide association studies in schizophrenia: recent advances, challenges and future perspective. Schizophr. Res. https://doi.org/10.1016/j.schres.2019.10.048 (2019).
Golov, A. K., Kondratyev, N. V., Kostyuk, G. P. & Golimbet, A. V. E. Novel approaches for identifying the molecular background of schizophrenia. Cells 9, https://doi.org/10.3390/cells9010246 (2020).
Weinberger, D. R. Thinking about schizophrenia in an era of genomic medicine. Am. J. Psychiatry 176, 12–20 (2019).
Zhuo, C. et al. The genomics of schizophrenia: shortcomings and solutions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 93, 71–76 (2019).
Hashimoto, K. Targeting of alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia and the use of auditory sensory gating as a translational biomarker. Curr. Pharm. Des. 21, 3797–3806 (2015).
de Leon, J. & Diaz, F. J. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophrenia Res. 76, 135–157 (2005).
Picciotto, M. R. & Zoli, M. Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Front. Biosci. 13, 492–504 (2008).
Featherstone, R. E. et al. Nicotine receptor subtype-specific effects on auditory evoked oscillations and potentials. PLoS ONE 7, e39775 (2012).
Freedman, R. alpha7-nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Annu. Rev. Med. 65, 245–261 (2014).
Freedman, R., Adams, C. E. & Leonard, S. The alpha7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J. Chem. Neuroanat. 20, 299–306 (2000).
Hajos, M. et al. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J. Pharmacol. Exp. Ther 312, 1213–1222 (2005).
Olincy, A. & Stevens, K. E. Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem. Pharm. 74, 1192–1201 (2007).
Young, J. W. & Geyer, M. A. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem. Pharmacol. 86, 1122–1132 (2013).
Smucny, J. & Tregellas, J. R. Targeting neuronal dysfunction in schizophrenia with nicotine: evidence from neurophysiology to neuroimaging. J Psychopharmacol. 269881117705071, https://doi.org/10.1177/0269881117705071 (2017).
Weng, P. H. et al. CHRNA7 polymorphisms and dement risk: interactions with apolipoprotein epsilon4 and cigarette smoking. Sci. Rep. 6, 27231 (2016).
Carruthers, S. P., Gurvich, C. T. & Rossell, S. L. The muscarinic system, cognition and schizophrenia. Neurosci. Biobehav. Rev. 55, 393–402 (2015).
Felder, C. C. et al. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 68, 2605–2613 (2001).
McKinzie, D. L. & Bymaster, F. P. Muscarinic mechanisms in psychotic disorders. Handb. Exp. Pharmacol, 233–265, https://doi.org/10.1007/978-3-642-25758-2_9 (2012).
Raedler, T. J., Bymaster, F. P., Tandon, R., Copolov, D. & Dean, B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 12, 232–246 (2007).
Eglen, R. M. Muscarinic receptor subtype pharmacology and physiology. Prog. Med. Chem. 43, 105–136 (2005).
Eglen, R. M. Muscarinic receptor subtypes in neuronal and non-neuronal cholinergic function. Auton. Autacoid Pharmacol. 26, 219–233 (2006).
Levey, A. I. Immunological localization of m1–m5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci. 52, 441–448 (1993).
Levey, A. I., Edmunds, S. M., Heilman, C. J., Desmond, T. J. & Frey, K. A. Localization of muscarinic m3 receptor protein and M3 receptor binding in rat brain. Neuroscience 63, 207–221 (1994).
Levey, A. I., Kitt, C. A., Simonds, W. F., Price, D. L. & Brann, M. R. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 11, 3218–3226 (1991).
Barak, S. Modeling cholinergic aspects of schizophrenia: focus on the antimuscarinic syndrome. Behav. Brain Res. 204, 335–351 (2009).
Cancelli, I., Beltrame, M., D’Anna, L., Gigli, G. L. & Valente, M. Drugs with anticholinergic properties: a potential risk factor for psychosis onset in Alzheimer’s disease? Expert Opin. Drug Saf. 8, 549–557 (2009).
Cancelli, I., Valentinis, L., Merlino, G., Valente, M. & Gigli, G. L. Drugs with anticholinergic properties as a risk factor for psychosis in patients affected by Alzheimer’s disease. Clin. Pharm. Ther. 84, 63–68 (2008).
Janowsky, D. S., el-Yousef, M. K., Davis, J. M. & Sekerke, H. J. A cholinergic–adrenergic hypothesis of mania and depression. Lancet 2, 632–635 (1972).
Gershon, S. & Shaw, F. H. Psychiatric sequelae of chronic exposure to organophosphorus insecticides. Lancet 1, 1371–1374 (1961).
Tandon, R. & Greden, J. F. Cholinergic hyperactivity and negative schizophrenic symptoms. A model of cholinergic/dopaminergic interactions in schizophrenia. Arch. Gen. Psychiatry 46, 745–753 (1989).
Giachetti, A., Giraldo, E., Ladinsky, H. & Montagna, E. Binding and functional profiles of the selective M1 muscarinic receptor antagonists trihexyphenidyl and dicyclomine. Br. J. Pharm. 89, 83–90 (1986).
Smith, J. M. Abuse of the antiparkinson drugs: a review of the literature. J. Clin. Psychiatry 41, 351–354 (1980).
Bakker, G. et al. Relationship between muscarinic M1 receptor binding and cognition in medication-free subjects with psychosis. NeuroImage Clin. 18, 713–719 (2018).
Newman, E. L., Gupta, K., Climer, J. R., Monaghan, C. K. & Hasselmo, M. E. Cholinergic modulation of cognitive processing: insights drawn from computational models. Front. Behav. Neurosci. 6, 24 (2012).
Russo, S. J. & Nestler, E. J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625 (2013).
Faure, P., Tolu, S., Valverde, S. & Naudé, J. Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neuroscience 282, 86–100 (2014).
Zhao-Shea, R. et al. Nicotine-mediated activation of dopaminergic neurons in distinct regions of the ventral tegmental area. Neuropsychopharmacology 36, 1021–1032 (2011).
Ropacki, S. A. & Jeste, D. V. Epidemiology of and risk factors for psychosis of Alzheimer’s disease: a review of 55 studies published from 1990 to 2003. Am. J. Psychiatry 162, 2022–2030 (2005).
Koppel, J. et al. Psychosis in Alzheimer’s disease is associated with frontal metabolic impairment and accelerated decline in working memory: findings from the Alzheimer’s Disease Neuroimaging Initiative. Am. J. Geriatr. Psychiatry 22, 698–707 (2014).
Reeves, S. J., Gould, R. L., Powell, J. F. & Howard, R. J. Origins of delusions in Alzheimer’s disease. Neurosci. Biobehav. Rev. 36, 2274–2287 (2012).
Garcia-Alloza, M. et al. Cholinergic–serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer’s disease. Neuropsychologia 43, 442–449 (2005).
Martin, L. F. & Freedman, R. Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int. Rev. Neurobiol. 78, 225–246 (2007).
Martin, L. F., Kem, W. R. & Freedman, R. Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacology 174, 54–64 (2004).
Gault, J. et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics 52, 173–185 (1998).
Prado, V. F., Janickova, H., Al-Onaizi, M. A. & Prado, M. A. Cholinergic circuits in cognitive flexibility. Neuroscience 345, 130–141 (2017).
Hampel, H. et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933 (2018).
Schmitz, T. W. & Nathan Spreng, R. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 7, 13249 (2016).
Threlfell, S. & Cragg, S. J. Dopamine signaling in dorsal versus ventral striatum: the dynamic role of cholinergic interneurons. Front. Syst. Neurosci. 5, 11 (2011).
Cummings, J. L. & Kaufer, D. Neuropsychiatric aspects of Alzheimer’s disease: the cholinergic hypothesis revisited. Neurology 47, 876–883 (1996).
Ballard, C. et al. Delusions associated with elevated muscarinic binding in dementia with Lewy bodies. Ann. Neurol. 48, 868–876 (2000).
Raedler, T. J. et al. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am. J. Psychiatry 160, 118–127 (2003).
Byun, N. E. et al. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology 39, 1578–1593 (2014).
Lai, M. K. et al. Psychosis of Alzheimer’s disease is associated with elevated muscarinic M2 binding in the cortex. Neurology 57, 805–811 (2001).
Vakalopoulos, C. Neuropharmacology of cognition and memory: a unifying theory of neuromodulator imbalance in psychiatry and amnesia. Med. Hypotheses 66, 394–431 (2006).
Cummings, J. L., Mackell, J. & Kaufer, D. Behavioral effects of current Alzheimer’s disease treatments: a descriptive review. Alzheimers Dement. 4, 49–60 (2008).
Figiel, G. & Sadowsky, C. A systematic review of the effectiveness of rivastigmine for the treatment of behavioral disturbances in dementia and other neurological disorders. Curr. Med. Res. Opin. 24, 157–166 (2008).
Millan, M. J. et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat. Rev. Drug Discov. 11, 141–168 (2012).
Bronstein, M. V., Pennycook, G., Joormann, J., Corlett, P. R. & Cannon, T. D. Dual-process theory, conflict processing, and delusional belief. Clin. Psychol. Rev. 72, 101748 (2019).
Mehl, S. et al. Theory of mind, emotion recognition, delusions and the quality of the therapeutic relationship in patients with psychosis—a secondary analysis of a randomized-controlled therapy trial. BMC Psychiatry 20, 59 (2020).
Breese, C. R. et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 23, 351–364 (2000).
Durany, N. et al. Human post-mortem striatal alpha4beta2 nicotinic acetylcholine receptor density in schizophrenia and Parkinson’s syndrome. Neurosci. Lett. 287, 109–112 (2000).
Zhang, T. et al. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J. Neurosci. 29, 4035–4043 (2009).
Holt, D. J. et al. Reduced density of cholinergic interneurons in the ventral striatum in schizophrenia: an in situ hybridization study. Biol. Psychiatry 58, 408–416 (2005).
Holt, D. J. et al. Evidence for a deficit in cholinergic interneurons in the striatum in schizophrenia. Neuroscience 94, 21–31 (1999).
Leslie, F. M., Mojica, C. Y. & Reynaga, D. D. Nicotinic receptors in addiction pathways. Mol. Pharm. 83, 753–758 (2013).
Esterlis, I. et al. In vivo evidence for beta2 nicotinic acetylcholine receptor subunit upregulation in smokers as compared with nonsmokers with schizophrenia. Biol. Psychiatry 76, 495–502 (2014).
Olincy, A. & Freedman, R. Nicotinic mechanisms in the treatment of psychotic disorders: a focus on the alpha7 nicotinic receptor. Handb. Exp. Pharmacol. 211–232, https://doi.org/10.1007/978-3-642-25758-2_8 (2012).
Threlfell, S. et al. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64 (2012).
McCutcheon, R., Beck, K., Jauhar, S. & Howes, O. D. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophrenia Bull. 44, 1301–1311 (2018).
McCutcheon, R. A., Abi-Dargham, A. & Howes, O. D. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. https://doi.org/10.1016/j.tins.2018.12.004 (2019).
Abudukeyoumu, N., Hernandez-Flores, T., Garcia-Munoz, M. & Arbuthnott, G. W. Cholinergic modulation of striatal microcircuits. Eur. J. Neurosci. 49, 604–622 (2019).
Brimblecombe, K. R. & Cragg, S. J. The striosome and matrix compartments of the striatum: a path through the labyrinth from neurochemistry toward function. ACS Chem. Neurosci. 8, 235–242 (2017).
Graybiel, A. M. & Ragsdale, C. W. Jr. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl Acad. Sci. USA 75, 5723–5726 (1978).
Lim, S. A., Kang, U. J. & McGehee, D. S. Striatal cholinergic interneuron regulation and circuit effects. Front. Synaptic Neurosci. 6, 22, (2014).
Bernacer, J., Prensa, L. & Gimenez-Amaya, J. M. Cholinergic interneurons are differentially distributed in the human striatum. PLoS ONE 2, e1174 (2007).
Aliane, V., Perez, S., Bohren, Y., Deniau, J. M. & Kemel, M. L. Key role of striatal cholinergic interneurons in processes leading to arrest of motor stereotypies. Brain 134, 110–118 (2011).
Bordia, T., Perez, X. A., Heiss, J., Zhang, D. & Quik, M. Optogenetic activation of striatal cholinergic interneurons regulates l-dopa-induced dyskinesias. Neurobiol. Dis. 91, 47–58 (2016).
Dean, B. et al. The density of muscarinic M1 receptors is decreased in the caudate-putamen of subjects with schizophrenia. Mol. Psychiatry 1, 54–58 (1996).
Dawson, V. L., Dawson, T. M. & Wamsley, J. K. Muscarinic and dopaminergic receptor subtypes on striatal cholinergic interneurons. Brain Res. Bull. 25, 903–912 (1990).
Court, J. A. et al. Nicotine binding in human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease and in relation to neuroleptic medication. Neuroscience 98, 79–87 (2000).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Factor, S. A., McDonald, W. M. & Goldstein, F. C. The role of neurotransmitters in the development of Parkinson’s disease-related psychosis. Eur. J. Neurol. 24, 1244–1254 (2017).
Warren, N., O’Gorman, C., Hume, Z., Kisely, S. & Siskind, D. Delusions in Parkinson’s disease: a systematic review of published cases. Neuropsychol. Rev. 28, 310–316 (2018).
Factor, S. A. et al. Disease-related and genetic correlates of psychotic symptoms in Parkinson’s disease. Mov. Disord. 26, 2190–2195 (2011).
Janzen, J. et al. The pedunculopontine nucleus is related to visual hallucinations in Parkinson’s disease: preliminary results of a voxel-based morphometry study. J. Neurol. 259, 147–154 (2012).
Factor, S. A. et al. Cognitive correlates of hallucinations and delusions in Parkinson’s disease. J. Neurol. Sci. 347, 316–321 (2014).
Poletti, M. et al. Dopamine agonists and delusional jealousy in Parkinson’s disease: a cross-sectional prevalence study. Mov. Disord. 27, 1679–1682 (2012).
Stefanis, N. et al. Isolated delusional syndrome in Parkinson’s disease. Parkinsonism Relat. Disord. 16, 550–552 (2010).
Wolters, E. C. Intrinsic and extrinsic psychosis in Parkinson’s disease. J. Neurol. 248(Suppl. 3), III22–III27 (2001).
Hshieh, T. T., Fong, T. G., Marcantonio, E. R. & Inouye, S. K. Cholinergic deficiency hypothesis in delirium: a synthesis of current evidence. J. Gerontol. 63, 764–772 (2008).
Hijazi, Z., Lange, P., Watson, R. & Maier, A. B. The use of cerebral imaging for investigating delirium aetiology. Eur. J. Intern. Med. 52, 35–39 (2018).
Nitchingham, A., Kumar, V., Shenkin, S., Ferguson, K. J. & Caplan, G. A. A systematic review of neuroimaging in delirium: predictors, correlates and consequences. Int. J. Geriatr. Psychiatry 33, 1458–1478 (2018).
Salviati, M. et al. Capgras-like syndrome in a patient with an acute urinary tract infection. Neuropsychiatr. Dis. Treat. 9, 139–142 (2013).
Rocha, N. P. et al. The clinical picture of psychosis in manifest Huntington’s disease: a comprehensive analysis of the Enroll-HD database. Front. Neurol. 9, 930 (2018).
Paulsen, J. S., Ready, R. E., Hamilton, J. M., Mega, M. S. & Cummings, J. L. Neuropsychiatric aspects of Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 71, 310–314 (2001).
Duff, K. et al. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol. Psychiatry 62, 1341–1346 (2007).
Hoth, K. F. et al. Patients with Huntington’s disease have impaired awareness of cognitive, emotional, and functional abilities. J. Clin. Exp. Neuropsychol. 29, 365–376 (2007).
McColgan, P. & Tabrizi, S. J. Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34 (2018).
Yager, L. M., Garcia, A. F., Wunsch, A. M. & Ferguson, S. M. The ins and outs of the striatum: role in drug addiction. Neuroscience 301, 529–541 (2015).
Aronin, N., Kim, M., Laforet, G. & DiFiglia, M. Are there multiple pathways in the pathogenesis of Huntington’s disease? Philos. Trans. R. Soc. Lond. B 354, 995–1003 (1999).
Rub, U. et al. Huntington’s disease (HD): neurodegeneration of Brodmann’s primary visual area 17 (BA17). Brain Pathol. 25, 701–711 (2015).
Schonberger, S. J., Jezdic, D., Faull, R. L. & Cooper, G. J. Proteomic analysis of the human brain in Huntington’s disease indicates pathogenesis by molecular processes linked to other neurodegenerative diseases and to type-2 diabetes. J. Huntington’s Dis. 2, 89–99 (2013).
Waldvogel, H. J., Kim, E. H., Thu, D. C., Tippett, L. J. & Faull, R. L. New perspectives on the neuropathology in Huntington’s disease in the human brain and its relation to symptom variation. J. Huntington’s Dis. 1, 143–153 (2012).
Ferrante, R. J., Beal, M. F., Kowall, N. W., Richardson, E. P. Jr. & Martin, J. B. Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 411, 162–166 (1987).
Adams, R. A., Stephan, K. E., Brown, H. R., Frith, C. D. & Friston, K. J. The computational anatomy of psychosis. Front. Psychiatry 4, 47 (2013).
Fletcher, P. C. & Frith, C. D. Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat. Rev. Neurosci. 10, 48–58 (2009).
Seth, A. K. & Tsakiris, M. Being a beast machine: the somatic basis of selfhood. Trends Cogn. Sci. 22, 969–981 (2018).
Deperrois, N., Moiseeva, V. & Gutkin, B. Minimal circuit model of reward prediction error computations and effects of nicotinic modulations. Front. Neural Circuits 12, 116 (2018).
Moran, R. J. et al. Free energy, precision and learning: the role of cholinergic neuromodulation. J. Neurosci. 33, 8227–8236 (2013).
Vossel, S. et al. Cholinergic stimulation enhances Bayesian belief updating in the deployment of spatial attention. J. Neurosci. 34, 15735–15742 (2014).
Yu, A. J. & Dayan, P. Uncertainty, neuromodulation, and attention. Neuron 46, 681–692 (2005).
Yamanaka, K. et al. Roles of centromedian parafascicular nuclei of thalamus and cholinergic interneurons in the dorsal striatum in associative learning of environmental events. J. Neural Transm. 125, 501–513 (2018).
Goldberg, J. A. & Reynolds, J. N. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience 198, 27–43 (2011).
Wang, X. C., Du, X. X., Tian, Q. & Wang, J. Z. Correlation between choline signal intensity and acetylcholine level in different brain regions of rat. Neurochem. Res. 33, 814–819 (2008).
Shattuck, K. F. & VanMeter, J. W. Task-based changes in proton MR spectroscopy signal during configural working memory in human medial temporal lobe. J. Magn. Reson Imaging 47, 682–691 (2018).
Yu, A. J. & Dayan, P. Acetylcholine in cortical inference. Neural Netw. 15, 719–730 (2002).
Buchanan, R. W. et al. A randomized clinical trial of oxytocin or galantamine for the treatment of negative symptoms and cognitive impairments in people with schizophrenia. J. Clin. Psychopharmacol. 37, 394–400 (2017).
Ochoa, E. L. M. & Clark, E. Galantamine may improve attention and speech in schizophrenia. Hum. Psycopharmacol. 21, 127–128 (2006).
Pae, C. U. Role of the cholinesterase inhibitors in the treatment of schizophrenia. Expert Opin. Investig. Drugs 22, 293–298 (2013).
Shoja Shafti, S. & Azizi Khoei, A. Effectiveness of rivastigmine on positive, negative, and cognitive symptoms of schizophrenia: a double-blind clinical trial. Ther. Adv. Psychopharmacol. 6, 308–316 (2016).
Corlett, P. R. & Fletcher, P. C. The neurobiology of schizotypy: fronto-striatal prediction error signal correlates with delusion-like beliefs in healthy people. Neuropsychologia 50, 3612–3620 (2012).
Trytek, E. S., White, I. M., Schroeder, D. M., Heidenreich, B. A. & Rebec, G. V. Localization of motor- and nonmotor-related neurons within the matrix-striosome organization of rat striatum. Brain Res. 707, 221–227 (1996).
Canales, J. J. & Graybiel, A. M. A measure of striatal function predicts motor stereotypy. Nat. Neurosci. 3, 377–383 (2000).
Berridge, K. C., Aldridge, J. W., Houchard, K. R. & Zhuang, X. Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette’s. BMC Biol. 3, 4 (2005).
Groth, C. Tourette syndrome in a longitudinal perspective. Clinical course of tics and comorbidities, coexisting psychopathologies, phenotypes and predictors. Dan. Med. J. 65, B5465 (2018).
Attademo, L., De Giorgio, G., Quartesan, R. & Moretti, P. Schizophrenia and obsessive-compulsive disorder: from comorbidity to schizo-obsessive disorder. Riv. Psichiatr. 47, 106–115 (2012).
Frias, A. et al. Neuropsychological profile and treatment-related features among patients with comorbidity between schizophrenia spectrum disorder and obsessive-compulsive disorder: is there evidence for a “schizo-obsessive” subtype? Psychiatry Res. 220, 846–854 (2014).
Silver, A. A., Shytle, R. D. & Sanberg, P. R. Mecamylamine in Tourette’s syndrome: a two-year retrospective case study. J. Child Adolesc. Psychopharmacol. 10, 59–68 (2000).
Brimblecombe, K. R. & Cragg, S. J. Substance P weights striatal dopamine transmission differently within the striosome–matrix axis. J. Neurosci. 35, 9017–9023 (2015).
Inoue, R., Suzuki, T., Nishimura, K. & Miura, M. Nicotinic acetylcholine receptor-mediated GABAergic inputs to cholinergic interneurons in the striosomes and the matrix compartments of the mouse striatum. Neuropharmacology 105, 318–328 (2016).
Crittenden, J. R. et al. Striatal cholinergic interneurons modulate spike-timing in striosomes and matrix by an amphetamine-sensitive mechanism. Front. Neuroanat. 11, 20 (2017).
Graybiel, A. M. The basal ganglia and cognitive pattern generators. Schizophrenia Bull. 23, 459–469 (1997).
Adams, R. A., Napier, G., Roiser, J. P., Mathys, C. & Gilleen, J. Attractor-like dynamics in belief updating in schizophrenia. J. Neurosci. 38, 9471–9485 (2018).
Partridge, J. G., Apparsundaram, S., Gerhardt, G. A., Ronesi, J. & Lovinger, D. M. Nicotinic acetylcholine receptors interact with dopamine in induction of striatal long-term depression. J. Neurosci. 22, 2541–2549 (2002).
Atta, K., Forlenza, N., Gujski, M., Hashmi, S. & Isaac, G. Delusional misidentification syndromes: separate disorders or unusual presentations of existing DSM-IV categories? Psychiatry (Edgmont (Pa.: Townsh.)) 3, 56–61 (2006).
Darby, R. R., Laganiere, S., Pascual-Leone, A., Prasad, S. & Fox, M. D. Finding the imposter: brain connectivity of lesions causing delusional misidentifications. Brain 140, 497–507 (2017).
Kuo, H. Y. & Liu, F. C. Synaptic wiring of corticostriatal circuits in basal ganglia: insights into the pathogenesis of neuropsychiatric disorders. eNeuro 6, https://doi.org/10.1523/eneuro.0076-19.2019 (2019).
Corlett, P. R. Factor one, familiarity and frontal cortex: a challenge to the two-factor theory of delusions. Cogn. Neuropsychiatry 24, 165–177 (2019).
Haydar, S. N. & Dunlop, J. Neuronal nicotinic acetylcholine receptors—targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top. Med. Chem. 10, 144–152 (2010).
Cummings, J. L., Gorman, D. G. & Shapira, J. Physostigmine ameliorates the delusions of Alzheimer’s disease. Biol. Psychiatry 33, 536–541 (1993).
Wallace, T. L. & Porter, R. H. Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem. Pharmacol. 82, 891–903 (2011).
Perez Lloret, S., Peralta, M. C. & Barrantes, F. J. in Psychiatry and Neurosciences. Update 2016. Translational Research (eds Gargiulo, P. A. & Mesones Arroyo, H.) (Springer, 2017).
Perez-Lloret, S., Peralta, M. C. & Barrantes, F. J. Pharmacotherapies for Parkinson’s disease symptoms related to cholinergic degeneration. Expert Opin. Pharmacother. 17, 2405–2415 (2016).
Uteshev, V. V. The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur. J. Pharmacol. 727, 181–185 (2014).
Vallés, A. S., Borroni, M. V. & Barrantes, F. J. Targeting brain α7 nicotinic acetylcholine receptors in Alzheimer’s disease: rationale and current status. CNS Drugs 28, 975–987 (2014).
Bertrand, D. et al. Positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2–M3 segment. Mol. Pharm. 74, 1407–1416 (2008).
Bertrand, D. & Gopalakrishnan, M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharm. 74, 1155–1163 (2007).
Bertrand, D., Gopalakrishnan, M. & Donnelly-Roberts, D. Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers in basic research and clinical science-editorial perspective. Biochem. Pharm. 78, 657 (2009).
Nikiforuk, A., Kos, T., Potasiewicz, A. & Popik, P. Positive allosteric modulation of alpha 7 nicotinic acetylcholine receptors enhances recognition memory and cognitive flexibility in rats. Eur. Neuropsychopharmacol. 25, 1300–1313 (2015).
daCosta, C. J., Free, C. R., Corradi, J., Bouzat, C. & Sine, S. M. Single-channel and structural foundations of neuronal alpha7 acetylcholine receptor potentiation. J. Neurosci. 31, 13870–13879 (2011).
Potasiewicz, A., Nikiforuk, A., Holuj, M. & Popik, P. Stimulation of nicotinic acetylcholine alpha7 receptors rescue schizophrenia-like cognitive impairments in rats. J. Psychopharmacol. 31, 260–271 (2017).
Bender, A. M., Jones, C. K. & Lindsley, C. W. Classics in chemical neuroscience: xanomeline. ACS Chem. Neurosci. 8, 435–443 (2017).
Shekhar, A. et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 165, 1033–1039 (2008).
Bodick, N. C. et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473 (1997).
Nakano, K. Neural circuits and topographic organization of the basal ganglia and related regions. Brain Dev. 22(Suppl. 1), S5–S16 (2000).
Author information
Authors and Affiliations
Contributions
M.C., E.L.M.O., and F.J.B. conceived the idea, designed, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caton, M., Ochoa, E.L.M. & Barrantes, F.J. The role of nicotinic cholinergic neurotransmission in delusional thinking. npj Schizophr 6, 16 (2020). https://doi.org/10.1038/s41537-020-0105-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41537-020-0105-9
This article is cited by
-
Tremendous Fidelity of Vitamin D3 in Age-related Neurological Disorders
Molecular Neurobiology (2024)
-
Generation of Functional and Mature Sympathetic Neurons from Human Pluripotent Stem Cells via a Neuroepithelial Route
Journal of Molecular Neuroscience (2024)
-
Increased Metabolic Potential, Efficacy, and Safety of Emerging Treatments in Schizophrenia
CNS Drugs (2023)
-
Microstructural imaging and transcriptomics of the basal forebrain in first-episode psychosis
Translational Psychiatry (2022)
