
Abstract
Several lines of evidence suggest that components of the tumor microenvironment, specifically basement membrane and extracellular matrix proteins, influence drug sensitivities. We previously reported differential drug sensitivity of tumor cells localized adjacent to laminin-rich extracellular matrix in three-dimensional tumor spheroid cultures. To evaluate whether differential intra-tumor responses to targeted therapy occur in vivo, we examined the sensitivity of human epidermal growth factor receptor 2-positive tumors to lapatinib using a previously described ductal carcinoma in situ-like model characterized by tumor cell confinement within ductal structures surrounded by an organized basement membrane. Here we show that tumor cells localized to a ‘niche’ in the outer layer of the intraductal tumors adjacent to myoepithelial cells and basement membrane are resistant to lapatinib. We found that the pro-survival protein BCL2 is selectively induced in the niche-protected tumor cells following lapatinib treatment, and combined inhibition of HER2 and BCL-2/XL enhanced targeting of these residual tumor cells. Elimination of the niche-protected tumor cells was achieved with the HER2 antibody–drug conjugate T-DM1, which delivers a chemotherapeutic payload. Thus, these studies provide evidence that subpopulations of tumor cells within specific microenvironmental niches can adapt to inhibition of critical oncogenic pathways, and furthermore reveal effective strategies to eliminate these resistant subpopulations.
Similar content being viewed by others


Introduction
Extracellular matrix (ECM) proteins produced by diverse tumor types protect tumor cells from death in response to various agents.3,4,5,6 Work from our laboratory and others in three-dimensional (3D) culture systems has defined a protective role for ECM within the context of both normal7 and tumor1 cells. Using epithelial tumor cell lines cultured in reconstituted basement membrane (BM), we previously found that the outer, ECM-attached cells are resistant to multiple different drug therapies.1 ECM protection involved activation of an adaptive response program, including FOXO-dependent transcriptional and cap-independent translational induction of multiple receptor tyrosine kinases (RTKs) and pro-survival BCL2 family proteins.
To address whether a similar differential adaptive response is observed in vivo, we examined a tumor model that recapitulates the ECM-enveloped architecture of 3D spheroids by generating ductal carcinoma in situ (DCIS)-like tumors via intraductal injection of HER2+ SUM225 breast tumor cells.2 Since HER2+ DCIS accounts for 40–60% of all patient-related DCIS cases,8,9,10,11,12,13 this model represents one of the most relevant approaches to understand the biology of HER2+ DCIS and to evaluate HER2-targeted therapies within the context of pre-neoplastic breast cancer.
Results
To generate intraductal DCIS-like tumors, SUM225 breast tumor cells were transplanted via cleaved nipple injection into the mammary gland of 6–10-week-old female NOD/scid mice. The intraductal tumors recapitulated the histological architecture of human DCIS,2,14 with multiple layers of human epidermal growth factor receptor 2-positive (HER2+) tumor cells confined within a laminin-rich BM and a centralized necrotic core (Supplementary Fig. 1). SUM225 cells are resistant to trastuzumab, a HER2-targeted monoclonal antibody, but are sensitive to lapatinib, a small molecule dual RTK inhibitor of HER2 and epidermal growth factor receptor (EGFR).15,16,17 To examine the differential drug sensitivity of spatially distinct tumor cells in this model, female NOD/scid mice bearing HER2+ SUM225 DCIS-like tumors were randomized into two treatment groups: lapatinib monotherapy (200 mg/kg/day) or vehicle alone for a period of 5–10 days (n = 4–5 mice per group). Endpoint comparison of tumor sections by hematoxylin and eosin (H&E) analysis revealed that lapatinib treatment induced a reduction in the number of tumor cell layers, but did not eliminate the tumor cells closest to the BM (Fig. 1). The extent of tumor cell death and reduction of tumor mass varied in different regions of the tumor and in different experiments; however, under conditions in which a near complete reduction of the tumor cell layers was observed, the outer layer of tumor cells in contact with the myoepithelial cells and BM was spared (Fig. 1). HER2 IHC identified residual HER2+ tumor cells and indicated maintenance of HER2 status post-lapatinib (Fig. 1). Lapatinib treatment for a longer period, 21 days, resulted in similar preservation of these tumor cell populations (Fig. 1). Ki67 IHC revealed an overall reduction in the proliferation positive tumor cell population post-lapatinib (Supplementary Fig. 2). Interestingly, a subset of outer layer tumor cells maintain proliferative capacity post-treatment (Supplementary Fig. 2).

Preservation of niche-localized tumor cells post-lapatinib treatment. Representative H&E images of vehicle-treated (a–e) and 5-day or 10-day lapatinib-treated (f–j) SUM225 DCIS-like tumors. Note f, g, i were 10-day whereas h was 5-day lapatinib-treated. The lapatinib-treated tumor images represent the spectrum of responses observed, with the least significant reductions in the viable tumor cell content shown in (f). Note cell crypts (vacuole-like spaces throughout the cell layer) associated with the areas of cell death. More significant reduction in the viable tumor cell content is shown in (g, h), with maintenance of only the outermost layer of tumor cells adjacent to myoepithelial cells and BM in (h). HER2 immunostains confirm HER2 status among the residual tumor cell population (j). H&E (d, i) and serial section HER2 IHC (e, j) are presented. Note d, e, i, j represent longitudinal sections, whereas all other panels represent transverse sections, through the intraductal tumors. Comparison of vehicle (k–m) and 21-day lapatinib-treated (n–p) SUM225 tumors. Representative H&E (k, n) and serial section HER2 IHC (l, m) and (o, p) are presented. Arrowheads in l, o highlight the regions in m, p. Note preservation of the niche-localized HER2+ tumor cells post-long term lapatinib treatment (p). Scale bar, ~200 μm
We also examined another HER2+ tumor cell line, SUM190,18 that can generate intraductal tumors after intraductal transplantation. SUM190 maintain a non-invasive phenotype in vivo with histological similarities to SUM225. However, this model was uninformative with respect to differential drug sensitivity because both the outer and niche-associated tumor cells were insensitive to lapatinib, possibly due to a H1047R PIK3CA mutation, which has been shown to limit the effectiveness of lapatinib19 (Supplementary Fig. 3).
To explore potential mechanisms underlying the adaptation of SUM225 tumors to lapatinib treatment, we performed reverse phase protein arrays (RPPAs)20 on protein extracts prepared from vehicle-treated (n = 4) or lapatinib-treated (n = 5) tumor fragments (Fig. 2a). RPPA profile analysis confirmed that pathways downstream of HER2 and EGFR were inhibited post treatment (e.g., phosphorylation of EGFR, AKT and its targets, and mTOR targets). However, multiple RTKs, including HER2 and EGFR, were elevated in response to lapatinib treatment. Both observations are consistent with a previously described adaptive response mechanism mediated by release of feedback inhibition of pathways regulated by lapatinib1,21,22 and/or lapatinib-mediated stabilization and accumulation of HER2 protein.23,24,25

Lapatinib induces pro-survival BCL2 in the niche-protected tumor cells. a RPPA analysis of SUM225 vehicle and 10-day lapatinib-treated tumors. Proteins with significant differences (unpaired two-tail Welch’s t test p value < 0.05) between lapatinib-treated and vehicle-treated tumors are shown. Data are log2 transformed and median centered. Statistical analysis was performed in R v.3.2.2. The RPPA heatmap was generated in Cluster v.3.0 and Java TreeView v.1.1.1. b–g Matched tumor sections were assayed for BCL2 via IHC. Representative vehicle-treated and lapatinib-treated tumors from two independent experiments are presented. IHC assays confirmed the RPPA results and highlighted selective BCL2 induction within niche-localized tumor cells. h BCL2 levels were scored according to intensity as 0, 1+, 2+, or 3+ and summarized across multiple tumors from two independent experiments (Fisher’s exact test vehicle vs. 5-day lapatinib; p value = 0.0007145 and vehicle vs. 10-day lapatinib p value = 4.114e-05). Post lapatinib, BCL2 expression was largely localized within the outermost tumor cells. i–o Lapatinib treatment did not alter expression of the pro-survival protein BCL-XL. Note SUM225 tumors express moderate to high BCL-XL levels with or without treatment and throughout all viable cells (Fisher’s exact test vehicle vs. 5-day lapatinib; p value = 1.083e-05 and vehicle vs. 10-day lapatinib p value = 4.114e-05). Fisher’s exact tests were performed in R v.3.2.2. Arrowheads (b, e, i, l) highlight regions magnified in subsequent panels. Scale bar, ~200 μm
RPPA analysis also revealed an induction of BCL2 following lapatinib treatment (Fig. 2). This result is consistent with our previous in vitro data identifying BCL2 as a component of the adaptive response to PI3K/mTOR inhibition in ovarian cancer cells.1 To determine whether BCL2 was induced throughout the entire tumor or specifically enriched in the protected outer cells, we performed BCL2 IHC analysis on matched tumor tissue sections (Fig. 2). Blinded pathological assessment confirmed BCL2 induction following lapatinib treatment, and revealed that it selectively occurred in the outermost, lapatinib-resistant cells of the intraductal tumors (Fig. 2 and Supplementary Fig. 4). BCL2 induction was not coupled to estrogen receptor expression,26 as SUM225 tumors maintained an ER-negative phenotype (Supplementary Fig. 5).27 In contrast to BCL2, BCL-XL did not exhibit a similar BM-restricted pattern nor a notable change in expression level in response to lapatinib treatment (Fig. 2). The levels of BCL-XL were elevated throughout the entire tumor in lapatinib-treated tumors (Fig. 2).
To examine the functional relevance of BCL2 induction in the BM-localized tumor cells, we examined the effects of lapatinib treatment in combination with the dual BCL-2/XL inhibitor ABT-737.28 Mice bearing SUM225 DCIS-like tumors (n = 4–5 mice per group) were pre-treated with either vehicle or lapatinib [200 mg/kg/day per os (p.o.)] for 10 days followed by 5 days of either vehicle or lapatinib plus ABT-737 [70 mg/kg/day intraperitoneally (i.p.)]. Analyses of the tissue sections by H&E in parallel with HER2 IHC indicated that tumors treated with ABT-737 alone were indistinguishable from matched vehicle-treated tumors (Fig. 3). The combination treatment resulted in either greater reduction of the tumor cell layer or complete elimination of the residual tumor cells, including the outer layer of cells proximal to the basement membrane (Fig. 3). Quantification of these features revealed an overall increase in the loss of cells at the BM zone after treatment with lapatinib (Fig. 3s). ABT-737 treatment induced thrombocytopenia, a marker of ABT-737 pharmacodynamics in vivo, confirming on-target action in these experiments (Supplementary Fig. 6).29 Although the 5-day combination treatment did not eradicate the entire tumor, the enhancement in cell death indicates that BCL2 family proteins contribute to the adaptive survival response to lapatinib. Longer-term combination treatment was not feasible due to weight loss exceeding AAALAC limits (Supplementary Fig. 6).

Inhibition of BCL-2/XL in combination with lapatinib treatment eliminates a subset of the niche-localized tumor cells. Representative H&E low magnification (a–f) and high magnification (g–l) images are presented to compare treatment groups. Serial sections were subjected to HER2 IHC in order to visualize tumor cells. The matched HER2 stains are presented alongside the H&E stained sections (m–r). Arrowheads (a–f) highlight regions presented in g-l. Note multiple viable HER2+ tumor cells (g–i, k). Note complete elimination of HER2+ tumor cells (j, arrowhead) in response to lapatinib plus ABT-737 (dual BCL-2/XL inhibitor). Lapatinib plus ABT-199 (BCL2-selective inhibitor) treatment did not cause elimination of niche-localized cell populations (l). Graph (s) shows the percent loss of the outer niche-localized cells (clearance, quantified as described in the Methods) in multiple tumors from two independent experiments (Mann–Whitney test lapatinib vs. lapatinib + ABT-737; p value = 0.0357 one-tail and p value = 0.0715 two-tail). Each line represents the median. Scale bar, ~200 μm
To address whether selective inhibition of BCL2 in combination with HER2 blockade would induce a similar response, we examined the consequences of lapatinib treatment in combination with the BCL2-specific inhibitor ABT-199.29 ABT-199 treatment did not induce thrombocytopenia, which was consistent with BCL2-specific inhibition29 (Supplementary Fig. 6). Notably, the combination of lapatinib and ABT-199 did not significantly enhance the effects of lapatinib monotherapy (Fig. 3), highlighting the importance of ABT-737-induced BCL-XL inhibition as a critical aspect of combination treatment effectiveness. This result is consistent with our observation of constitutive BCL-XL expression within all cells of the tumor (Fig. 2).
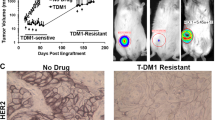
We also examined whether chemotherapeutic agents would provide better targeting and elimination of the protected tumor cell populations. As a means to deliver a cytotoxic agent directly to the HER2+ tumor cells, we utilized the HER2 ADC T-DM1. T-DM1 combines the anti-HER2 properties of trastuzumab with the anti-microtubule cytotoxic activities of DM1.30,31,32 Mice bearing SUM225 DCIS-like tumors were randomized into one of two treatment groups (n = 5 mice per group): single agent T-DM1 (10 mg/kg/1 × week i.p. 2 weeks) or vehicle. Analyses of the tissue sections by H&E in parallel with HER2 IHC indicated that T-DM1 treatment induced extensive cell death within the inner tumor cell layers as well as large sections of the outer, protected tumor cell population (Fig. 4). Quantification of these features revealed an overall enrichment of cell death at the BM zone (Fig. 4g). These results highlight T-DM1 as a means to eliminate niche-protected tumor cells.

T-DM1 treatment eliminates the outer and inner tumor cells. Representative H&E stained sections from vehicle-treated (a) and T-DM1-treated (d) SUM225 tumors are presented alongside serial section HER2 IHC (b, c) and (e, f). Arrowheads in b, e highlight the tumor nests that are presented in c, f. Note the significant elimination of HER2+ tumor cells post-T-DM1 treatment (e, f). Note niche-localized, and non-localized, cells are ultrasensitive to T-DM1 treatment. Pathological assessment of clearance was performed as described in the methods (g). Data was summarized across five vehicle-treated and five T-DM1-treated SUM225 tumors (Mann–Whitney test vehicle vs. T-DM1; p value = 0.0040 one-tail and p value = 0.0079 two-tail). Each line represents the median. Scale bar, ~100 μm
Discussion
Here we identified a DCIS sub-population of niche-associated tumor cells, adjacent to myoepithelial cells and BM, that are resistant to lapatinib and induce BCL2 post treatment. We found that HER2 blockade in combination with a BCL-2/XL inhibitor reduced the niche-protected population. However, the most effective elimination of these cells was observed after treatment with the antibody-drug conjugate, T-DM1, which delivers cytotoxic DM1 to HER2+ tumor cells.
While the precise mechanism responsible for lapatinib resistance within niche-localized tumor cells isn’t known, we predict that the BM, synthesized and organized by the myoepithelial cells, may be the most critical component of the protective response. Our previous studies, which utilized 3D tumor spheroids in vitro cultured in the absence of myoepithelial cells or other cellular tumor microenvironment components, revealed a similar differential resistance of the outer, matrix-attached cells treated with PI3K and/or mTOR inhibitors.1 In the in vivo DCIS-like model presented here, BCL2 upregulation was clearly enriched specifically in the outer, niche-localized drug resistant tumor cell populations. The lack of BCL2 upregulation in the remainder of the tumor suggests that cells beyond the ‘niche’ lack the abilities to adapt in a similar manner.
Outside of HER2+ disease, BCL-2/XL inhibition in combination with tamoxifen has proven efficacy in patient-derived xenograft (PDX) models of ER+ breast cancer.33 BCL2 is a known ER responsive gene, rendering it an especially attractive target for ER+ breast tumors,34 which exhibit a strong correlation with BCL2 expression regardless of HER2 status.35,36,37 BCL-2/XL blockade in combination with docetaxel also has proven efficacy in PDX models of triple negative breast cancer.38 Unlike SUM225 tumor cells treated with lapatinib, niche-localized SUM225 tumor cells were sensitive to a chemotherapeutic agent. These results highlight T-DM1 treatment as a means to prevent the persistence of niche-localized drug resistant tumor cells in vivo.
The adaptive response demonstrated here could facilitate the acquisition of additional genetic alterations in proliferating tumor cells that may lead to the generation of stably resistant tumor cells that can survive and proliferate in an niche-independent fashion. This could represent one mechanism for tumor recurrence in patients treated with targeted therapies. Indeed, two independently generated tumor cell models of acquired lapatinib resistance have identified BCL2 overexpression as a component of the resistant phenotype26,39 and suggest that BCL2 upregulation via the adaptive response (de novo resistance) could be maintained by stable, inherited alterations that lead to long-term acquired resistance. In line with this evidence, a recent report from a substantially large patient cohort supports reclassification of patients according to pCR or pCR + DCIS in correlation with disease-free survival.40
Our results highlight the potential for similar niches to confer protection in other tumor types. Since the BM is a notable component of prostate,41 lung3,42 and skin,43 tumor cells adjacent to BM could also be protected from targeted-therapies in this context. Indeed, in a mouse prostate non-invasive tumor model induced by PTEN deletion, outer tumor cells only were preserved in mice treated with mTOR inhibitors and residual tumor cells were BCL2-positive.44 The extent to which these protected niches are relevant to human tumors treated with targeted-therapies warrants future investigation. Although the relevant niche-associated components that confer protection in vivo remain to be defined, our results underscore the importance of microscopic evaluation of individual tumor cell responses to therapeutic agents in order to reveal spatially restricted heterogeneous drug responses. The identification and characterization of intra-tumoral protective microenvironmental niches may guide targeted therapeutic strategies to prevent drug resistance and disease recurrence in the future.
Materials and methods
Cell culture
HER2+ SUM225 and SUM190 breast tumor cells18,45 were maintained in complete media (Ham’s F12 supplemented with 5% heat-inactivated fetal bovine serum, 1 µg/ml hydrocortisone, 5 µg/ml insulin, 1% penicillin-streptomycin, 10 mM HEPES) by standard tissue culture practice at 37 °C and 5% CO2. SUM225 were kindly provided by Fariba Behbod (KU Medical Center). SUM190 were kindly provided by Dennis Slamon (UCLA).
Intraductal human-in-mouse tumor cell transplantation
Intraductal tumor cell transplants were performed as previously described.2 SUM225 or SUM190 cells were washed twice in PBS, trypsinized, counted and resuspended in complete media plus 0.1% trypan blue. Briefly, ~30 × 103 tumor cells in 3–5 µl were injected via cleaved nipple into the mammary gland of 6–10-week-old female NOD/scid mice. A 5 µl Hamilton syringe (75rn, G32, 2”, 90°) was used for all transplants. All animal procedures were carried out according to IACUC04004 and Harvard ARCM policy.
Tumor histology and IHC
Tumor tissue was harvested at the experimental endpoint and fixed in 3.7% paraformaldehyde (Boston BioProducts) or 10% neutral buffered formalin (Sigma). Tissue was processed for paraffin embedding, sectioning and H&E staining by the Harvard Rodent Histopathology core. Unstained sections were analyzed by immunohistochemical analysis according to standard protocol. For expanded details on immunohistochemistry methods and antibodies, please refer to the Supplementary Methods.
Drug treatment in vivo
For in vivo studies, lapatinib15,46,47,48 (GlaxoSmithKline) was administered p.o. at 200 mg/kg once per day. Lapatinib was prepared according to GlaxoSmithKline recommendations as a suspension in 0.5% hydroxypropylmethylcellulose (Dow Chemical K-15M) plus 0.1% Tween 80 (Sigma) in water. ABT-73728 (AbbVie) was administered i.p. at 70 mg/kg once per day. ABT-737 was prepared according to AbbVie recommendations in 30% propylene glycol (Sigma) plus 0.5% Tween 80 (Sigma) and 65% D5W (5% dextrose in water, Sigma) pH 3-4. ABT-19929 (AbbVie) was administered p.o. at 70 mg/kg once per day. ABT-199 was prepared according to AbbVie recommendations in 60% PHOSAL 50 PG (Lipoid) plus 30% polyethylene glycol 400 (Dow Chemical) and 10% ethanol. T-DM130 (Genentech) was administered i.p. at 10 mg/kg once per week. T-DM1 was prepared according to Genentech recommendations in sterile water for injection (GIBCO). Matched animals received vehicle alone in the same manner as drug-treated counterparts. All animals were weighed and randomized into groups before treatment. Either the average weight (average of the largest and smallest animals) or individual weights were used for dose calculations. Lapatinib treatment experiments were performed with or without a short-term food fast period. Mice were deprived of food for 2 h before the treatment and for 1 h after the treatment. Retro-orbital blood collection was performed on ABT-treated and vehicle-treated mice at the experimental endpoint. Mouse platelet counts were measured by Charles River Research Animal Diagnostic Services (Wilmington, MA).
Reverse phase protein arrays
Fresh tumor tissue was snap frozen in liquid nitrogen and stored at −80 °C. For protein lysates, the tumor fragments were pulverized over liquid nitrogen and resuspended in RPPA lysis buffer or were sonicated in RPPA lysis buffer (1% Triton X-100, 50 mM HEPES pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid, 100 mM NaF, 10 mM NaPPi, 10% glycerol, 1 mM Na3VO4 plus 1 mM phenylmethylsulfonyl fluoride and 10 μg/ml aprotinin or protease (Roche 11697498001) and phosphatase (Roche 04906837001) inhibitor tablets). Lysates were mixed well and clarified by centrifugation at 13000 rpm for 10 min at 4 °C. Supernatants were collected and protein was quantified by BCA assay (Pierce). Cellular proteins were denatured by 1% sodium dodecyl sulfate (SDS) (with β-mercaptoethanol) and diluted in five twofold serial dilutions in dilution buffer (lysis buffer containing 1% SDS). Serial diluted lysates were arrayed on nitrocellulose-coated slides (Grace Biolab) by an Aushon 2470 Arrayer (Aushon BioSystems). A total of 5808 array spots were arranged on each slide including the spots corresponding to positive and negative controls prepared from mixed cell lysates or dilution buffer, respectively. Each slide was probed with a validated primary antibody plus a biotin-conjugated secondary antibody. Only antibodies with a Pearson correlation coefficient between RPPA and Western blotting of greater than 0.7 were used in reverse phase protein array studies. Antibodies with a single or dominant band on Western blotting were further assessed by direct comparison to RPPA using cell lines with differential protein expression or modulated with ligands, inhibitors or siRNA for phospho-proteins or structural proteins, respectively. The signal obtained was amplified using a Dako Cytomation-catalyzed system (Dako) and visualized by 3,3'-diaminobenzidine colorimetric reaction. The slides were scanned, analyzed, and quantified using Array-Pro Analyzer software (MediaCybernetics) to generate spot intensity. Each dilution curve was fitted with a logistic model (“Supercurve Fitting” developed by the Department of Bioinformatics and Computational Biology, MD Anderson Cancer Center). The model fits a single curve using all the samples (i.e., dilution series) on a slide with the signal intensity as the response variable, and the dilution steps as the independent variable. The fitted curve is plotted with the signal intensities, both observed and fitted, on the y-axis and the log2-concentration of proteins on the x-axis for diagnostic purposes. The protein concentrations of each set of slides were then normalized by median polish, which was corrected across samples by the linear expression values using the median expression levels of all antibody experiments to calculate a loading correction factor for each sample.
Microscopy
H&E and IHC images were captured on the laboratory Nikon Eclipse E200 microscope equipped with an Idea color camera and the SPOT software package. H&E and IHC images were also captured, at the Nikon Imaging Center at Harvard Medical School, on the Nikon 80i upright microscope equipped with a PriorProScanII motorized stage and a Nikon Digital Slight DS Fi1 color camera. The NIS-Elements software package was used for image acquisition and analysis. H&E and IHC images were scanned, at the Neurobiology Imaging Facility at Harvard Medical School, on the Olympus VS120-S5 microscope equipped with a Hamamatsu ORCA-R2 color camera. The VS-ASW-FL software package was used for image analysis.
Quantification of clearance
Pathological assessment of clearance was performed by rodent pathologist Dr. Roderick Bronson, who was blinded to tumor treatment identities. On average, 30 DCIS-like structures were evaluated from multiple tumors per treatment group (Supplementary Tables 1 and 2). DCIS-like structures were scored according to the percent of niche-localized cells eliminated per structure (clearance). The total number of structures with greater than 10% clearance was graphed per tumor in Figs. 3 and 4.
Quantification of Ki67
Pathological assessment of Ki67 was performed by rodent pathologist Dr. Roderick Bronson, who was blinded to tumor treatment identities. Twenty-five 10× fields were evaluated from multiple vehicle-treated and lapatinib-treated tumors. Fields were scored according to the percent of Ki67+ cells (numerical values 0–100) and to the localization of Ki67+ cells (categorical values outer Ki67 positive or negative). The percent average Ki67+ cells per 10× field was graphed per tumor in Supplementary Fig. 2g. The percent of outer Ki67+ cells per 10× field was graphed per tumor in Supplementary Fig. 2h.
Statistics
Statistical analyses (except where noted) were performed using GraphPad Prism version 6 for MAC.
Change history
22 September 2017
A correction to this article has been published and is linked from the HTML version of this article.
References
Muranen, T. et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239 (2012).
Behbod, F. et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 11, R66 (2009).
Sethi, T. et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 5, 662–668 (1999).
Sherman-Baust, C. A. et al. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell 3, 377–386 (2003).
Uhm, J. H., Dooley, N. P., Kyritsis, A. P., Rao, J. S. & Gladson, C. L. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin. Cancer Res. 5, 1587–1594 (1999).
Pupa, S. M. et al. Regulation of breast cancer response to chemotherapy by fibulin-1. Cancer Res. 67, 4271–4277 (2007).
Weaver, V. M. et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2, 205–216 (2002).
Hoque, A. et al. Her-2/neu gene amplification in ductal carcinoma in situ of the breast. Cancer Epidemiol. Biomarkers Prev. 11, 587–590 (2002).
Karlsson, E. et al. Clonal alteration of breast cancer receptors between primary ductal carcinoma in situ (DCIS) and corresponding local events. Eur. J. Cancer 50, 517–524 (2014).
Somerville, J. E., Clarke, L. A. & Biggart, J. D. c-erbB-2 overexpression and histological type of in situ and invasive breast carcinoma. J. Clin. Pathol. 45, 16–20 (1992).
Allred, D. C. et al. Overexpression of HER-2/neu and its relationship with other prognostic factors change during the progression of in situ to invasive breast cancer. Hum. Pathol. 23, 974–979 (1992).
Schimmelpenning, H. et al. Expression of the c-erbB-2 proto-oncogene product and nuclear DNA content in benign and malignant human breast parenchyma. Virchows Arch. A Pathol. Anat. Histopathol. 420, 433–440 (1992).
Schimmelpenning, H. et al. Immunohistochemical c-erbB-2 protooncogene expression and nuclear DNA content in human mammary carcinoma in situ. Am. J. Clin. Pathol. 97, S48–S52 (1992).
Gupta, P. B. & Kuperwasser, C. Disease models of breast cancer. Drug Discov. Today 1, 9–16 (2004).
Konecny, G. E. et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 66, 1630–1639 (2006).
O’Brien, N. A. et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 9, 1489–1502 (2010).
Sambade, M. J. et al. Lapatinib in combination with radiation diminishes tumor regrowth in HER2+ and basal-like/EGFR+ breast tumor xenografts. Int. J. Radiat. Oncol. Biol. Phys. 77, 575–581 (2010).
Barnabas, N. & Cohen, D. Phenotypic and molecular characterization of MCF10DCIS and SUM breast cancer cell lines. Int. J. Breast Cancer 2013, 872743 (2013).
Rexer, B. N., Chanthaphaychith, S., Dahlman, K. & Arteaga, C. L. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 16, R9 (2014).
Tibes, R. et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 5, 2512–2521 (2006).
Chandarlapaty, S. et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71 (2011).
Stuhlmiller, T. J. et al. Inhibition of Lapatinib-induced Kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404 (2015).
Scaltriti, M. et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28, 803–814 (2009).
Maruyama, T. et al. Lapatinib enhances herceptin-mediated antibody-dependent cellular cytotoxicity by up-regulation of cell surface HER2 expression. Anticancer Res. 31, 2999–3005 (2011).
Mimura, K. et al. Lapatinib inhibits receptor phosphorylation and cell growth and enhances antibody-dependent cellular cytotoxicity of EGFR- and HER2-overexpressing esophageal cancer cell lines. Int. J. Cancer 129, 2408–2416 (2011).
Wang, Y. C. et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 13, R121 (2011).
Bayliss, J., Hilger, A., Vishnu, P., Diehl, K. & El-Ashry, D. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clin. Cancer Res. 13, 7029–7036 (2007).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
Lewis Phillips, G. D. et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290 (2008).
Barok, M., Tanner, M., Koninki, K. & Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 13, R46 (2011).
Junttila, T. T., Li, G., Parsons, K., Phillips, G. L. & Sliwkowski, M. X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 128, 347–356 (2011).
Vaillant, F. et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 24, 120–129 (2013).
Perillo, B., Sasso, A., Abbondanza, C. & Palumbo, G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol. Cell. Biol. 20, 2890–2901 (2000).
Silvestrini, R. et al. The Bcl-2 protein: a prognostic indicator strongly related to p53 protein in lymph node-negative breast cancer patients. J. Natl. Cancer Inst. 86, 499–504 (1994).
Leek, R. D., Kaklamanis, L., Pezzella, F., Gatter, K. C. & Harris, A. L. bcl-2 in normal human breast and carcinoma, association with oestrogen receptor-positive, epidermal growth factor receptor-negative tumours and in situ cancer. Br. J. Cancer 69, 135–139 (1994).
Bhargava, V., Kell, D. L., van de Rijn, M. & Warnke, R. A. Bcl-2 immunoreactivity in breast carcinoma correlates with hormone receptor positivity. Am. J. Pathol. 145, 535–540 (1994).
Oakes, S. R. et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 109, 2766–2771 (2012).
Xia, W. et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc. Natl. Acad. Sci. USA 103, 7795–7800 (2006).
von Minckwitz, G. et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 30, 1796–1804 (2012).
Nagle, R. B. Role of the extracellular matrix in prostate carcinogenesis. J. Cell Biochem. 91, 36–40 (2004).
Goto, K. et al. Detection of early invasion on the basis of basement membrane destruction in small adenocarcinomas of the lung and its clinical implications. Mod. Pathol. 14, 1237–1245 (2001).
Watt, F. M. & Fujiwara, H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb Perspect. Biol. 3, a005124 (2011).
Majumder, P. K. et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 10, 594–601 (2004).
Neve, R. M. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 (2006).
Rusnak, D. W. et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 1, 85–94 (2001).
Rusnak, D. W. et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 40, 580–594 (2007).
Xia, W. et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 21, 6255–6263 (2002).
Acknowledgements
We would like to acknowledge Aleksandr Vagodny (Harvard Cell Biology) for technical assistance with the mouse experiments and the IHC. We would like to acknowledge Tona Gilmer (GlaxoSmithKline) for helpful insight on the proper usage of lapatinib throughout our experiments. We would also like to acknowledge Joel Leverson (AbbVie) and Deepak Sampath (Genentech) for helpful insight on the proper usage of ABT-737, ABT-199 and T-DM1 in our experiments. We are grateful for the expert histological assistance provided by Li Zhang (Harvard Pathology). We would like to thank Yiling Lu and the MD Anderson Cancer Center RPPA Core Facility (supported by NCI CA16672). We also thank Angie Martinez Gakidis for critical review and editing of the manuscript. We would also like to thank the Harvard Medical School Nikon Imaging Center and Neurobiology Imaging Facility (supported in part by NINDS P30 NS072030). Our work was supported by a Department of Defense Breast Cancer Research Fellowship Award W81XWH-11-1-0572 (J.J.Z.), an SU2C AACR Translational Cancer Research Award (J.S.B.), and the Breast Cancer Research Foundation (J.S.B.).
Author information
Authors and Affiliations
Contributions
J.J.Z. performed all in vivo mouse experiments and IHC tests on the tumor samples (with technical assistance from Aleksandr Vagodny) and drafted the manuscript. R.T.B. performed blinded analysis and provided expert insight on all mouse histology. L.M.S. performed statistical analysis and evaluation of RPPA data. G.B.M. performed the RPPA studies and discussed the results. J.S.B. participated in the planning and interpretation of experiments and in the preparation of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Change History
A correction to this article has been published and is linked from the HTML version of this article.
An erratum to this article is available at https://doi.org/10.1038/s41523-017-0031-9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zoeller, J.J., Bronson, R.T., Selfors, L.M. et al. Niche-localized tumor cells are protected from HER2-targeted therapy via upregulation of an anti-apoptotic program in vivo. npj Breast Cancer 3, 18 (2017). https://doi.org/10.1038/s41523-017-0020-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-017-0020-z
This article is cited by
-
Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies
Signal Transduction and Targeted Therapy (2023)
-
The importance of being CAFs (in cancer resistance to targeted therapies)
Journal of Experimental & Clinical Cancer Research (2022)
-
Ibrutinib reverses IL-6-induced osimertinib resistance through inhibition of Laminin α5/FAK signaling
Communications Biology (2022)
-
Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer
Nature Reviews Cancer (2022)
-
A SNAI2-PEAK1-INHBA stromal axis drives progression and lapatinib resistance in HER2-positive breast cancer by supporting subpopulations of tumor cells positive for antiapoptotic and stress signaling markers
Oncogene (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}