
Abstract
Metallic tungsten disulfide (WS2) monolayers have been demonstrated as promising electrocatalysts for hydrogen evolution reaction (HER) induced by the high intrinsic conductivity, however, the key challenges to maximize the catalytic activity are achieving the metallic WS2 with high concentration and increasing the density of the active sites. In this work, single-atom-V catalysts (V SACs) substitutions in 1T-WS2 monolayers (91% phase purity) are fabricated to significantly enhance the HER performance via a one-step chemical vapor deposition strategy. Atomic-resolution scanning transmission electron microscopy (STEM) imaging together with Raman spectroscopy confirm the atomic dispersion of V species on the 1T-WS2 monolayers instead of energetically favorable 2H-WS2 monolayers. The growth mechanism of V SACs@1T-WS2 monolayers is experimentally and theoretically demonstrated. Density functional theory (DFT) calculations demonstrate that the activated V-atom sites play vital important role in enhancing the HER activity. In this work, it opens a novel path to directly synthesize atomically dispersed single-metal catalysts on metastable materials as efficient and robust electrocatalysts.
Similar content being viewed by others

Introduction
Hydrogen fuel generation from water splitting is one of the most promising ways to replace conventional fossil fuels and solve the energy crisis1,2. Recently, various strategies have been developed to realize the highly efficient catalysts for hydrogen evolution reaction (HER), including semiconductor-based photocatalytic HER (polymer g-C3N43, Ag/semiconductor4, etc.), photoelectrochemically catalytic HER5, and metal-based electrocatalytic HER (metal sulfides1,6,7, metal carbides8,9, etc.). Commercially, noble metals from the Pt group are utilized to reduce the overpotential of HER and boost the kinetics with unrivaled activities, however, they usually suffer from scarcity, high-cost, and long-term instability. Hence, it is highly desirable to explore robust and efficient HER alternative catalysts with earth-abundant elements to realize the hydrogen economy.
Transition metal dichalcogenides (TMDs) from Group VI elements have recently kindled tremendous investigation as efficient Pt substitutes for HER catalysis because of the catalytically active S atoms on edge sites10. Unfortunately, the high proportion of inactive basal plane of MX2 (M = Mo or W, X = S or Se) significantly limits the catalytic performance because of the low electronic transfer capability, leading to the sluggish electrocatalytic kinetics1. Two key factors are worthy of being considered to maximize HER activity. One is to increase the metallic phase proportion of MX2, thus improving the intrinsic conductivity of MX2 and boosting HER activity2,6,11. However, it is still challenging to directly synthesize a highly pure metallic phase, especially for 1T-WS2, owing to the highest formation energy of 1T-WS2 (0.89 eV per formula) among all the polymorphs of MX2 (Supplementary Fig. 1)12. Recently, the phase-engineered syntheses of metallic MX2 from the 2H phase have been widely developed via wet-chemistry or exfoliated methods, however, the as-produced metallic MX2 domains were usually found with 1T(1T′)/2H mixed-phase and unstable after long-time air-exposure1,2,6,7,13,14,15,16,17,18,19,20,21,22. The other important factor is increasing the density of active sites of MX2. The field of single atomic catalysts (SACs) comprising isolated metal atoms on the varied supports gives new opportunities for the development of MX2 with increased active sites due to the high atom utilization of SACs23,24,25. However, to the best of our knowledge, the reports on the direct one-step vapor-phase synthesis of SACs on the highly pure metallic MX2 have not been explored.
Here, we show highly dispersed single vanadium atoms on the 1T-WS2 monolayers (denoted as V SACs@1T-WS2), which are synthesized through a one-step chemical vapor deposition (CVD) via controlling the introduction of VCl3. By using VCl3 as the co-precursor, the metallic tungsten disulfide (WS2) monolayers show an ultrahigh 1T concentration of 91%, which is the highest 1T ratio achieved by CVD so far (Supplementary Table 1). Remarkably, the V SACs@1T-WS2 monolayers show superior HER activity comparable to their 2H counterparts, with a low Tafel slope of 61 mV/dec and high turnover frequency (TOF) of 3.01 s−1 at 100 mV, and a remarkable long-term catalytic stability. We also demonstrate that the highly active single-atom V sites play a vital role in enhancing the HER activity of intrinsic 1T-WS2 monolayers.
Results
Characterization of V SACs@1T-WS2 monolayers
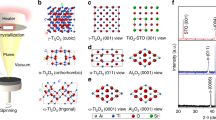
Figure 1 shows the scheme for the growth of V SACs@1T-WS2 monolayers using WO3, sulphur, and VCl3 as the co-precursors. For comparison, 2H-WS2 monolayers were prepared without using the VCl3 co-precursor at the same condition and the growth process has been reported elsewhere26,27,28,29,30,31,32,33. Strikingly, a black film of V2O3 (Fig. 2a and Supplementary Fig. 2) was observed on the sapphire surface during the V SACs@1T-WS2 monolayers growth, while no V2O3 film appeared for the 2H-WS2 monolayers growth. The optical micrographs of fresh sapphire and V2O3 film were also provided here for comparison, as seen in Supplementary Fig. 3a, b. In sharp contrast to the triangular morphology of 2H-WS2 (Fig. 2b), the V SACs@1T-WS2 monolayers show a uniformly circular morphology with a lateral size of 30 μm (Fig. 2b and Supplementary Fig. 3c).

I V2O3 formation in the early growth stage. II V SACs@1T-WS2 monolayers formed on the V2O3 film.

a Pictures of different materials formed on the sapphire substrates. Up: V2O3 film; Middle: V SACs@1T-WS2/V2O3 film; down: 2H-WS2. b Optical micrographs of as-grown V SACs@1T-WS2 (up) and 2H-WS2 (down) grown by CVD. c High-resolution HAADF-STEM image for the V SACs@1T-WS2. Inset: the corresponding fast Fourier transform (FFT) of (c). d Left: zoom-in high-resolution STEM image for the V SACs@1T-WS2 indicated by (c). Right: the schematic model of V SACs@1T-WS2. The blue spheres, yellow spheres, and red spheres represent the W atoms, S atoms, and V atoms, respectively. e EELS spectrum of vanadium element in the V SACs@1T-WS2 monolayer. f Intensity profiles of the pink dashed arrow indicated by Fig. 1d. g Low-resolution cross-section HAADF-STEM image of V SACs@1T-WS2/V2O3 film on the sapphire substrate. Inset: the interface between V2O3 and sapphire substrate. h High-resolution HAADF-STEM image of V SACs@1T-WS2 on the V2O3 film; scale bars: (b) up: 10 µm, down: 5 µm; (c) 1 nm, inset: 5 1/nm; (d) 0.5 nm; (g) 20 nm, inset: 1 nm; (h) 1 nm.
The atomic structure of V SACs@1T-WS2 monolayers was investigated by the aberration-corrected STEM, as provided in Fig. 2c, d, showing that the evident Z-contrast intensity sites are strongly dependent on the atomic weight. Figure 2d shows a magnified image of the region outlined by a white dashed rectangle in Fig. 2c. Both the STEM images display a hexagonal packing along [001] zone axis which is usually observed in monolayered 2H-WS2 (Supplementary Fig. 4a), as highlighted by the fast Fourier transform (FFT, Fig. 2c inset), implying no 1T′ reconstruction occurred1,18. In particular, only W atoms are identified because of a much lower atomic number of S compared with W, and the invisible S atoms were also illustrated in the reported metallic WS2 crystals1,16,22. In addition to the invisible S atoms, the atomic positions where W atoms are replaced by V atoms can also be obscurely seen due to the significantly reduced contrast at the W atomic sites (marked by the white dashed circles). The electron energy-loss spectroscopy (EELS) of V SACs@1T-WS2 in Fig. 2e shows two major features of V-L2 (524.05 eV) and V-L3 (517.03 eV) peaks assigned to V4+, affirming the V substitutions in the 1T-WS2 layer34. The EELS of S and O spectra in Supplementary Fig. 5 were shown to further reveal the presence of S and absence of O in the transferred 1T samples, excluding the V signals from V-based oxidations. The schematic atomic structure of V SACs@1T-WS2 is depicted in Fig. 2d (right), in which we replace W atoms (blue spheres) of WS2 with V atoms (red spheres) to illustrate isolated V atoms in 1T-WS2. Moreover, the W–S–S intensity sequence in Fig. 2f (from the pink dashed arrow in Fig. 2d) is also indicative of the 1T phase of WS2 due to a misaligned top and bottom S atom. In contrast, for the 2H-WS2 in Supplementary Fig. 4b, the S atoms at the top and bottom sublayers are overlapped, leading to an alternating W and S intensity pattern. In addition, W–S–S–V–S–S–W and W–V–W intensity profile sequences from both experimental and simulated STEM images are also achieved to verify the V atoms replacement at W sites (see details in Supplementary Figs. 6 and 7). It is anticipated that the V and S atoms could become visible if the 1T phase is transformed into the 2H phase, which has been shown in a pre-published literature35. To further confirm the V-atom concentration in the 1T-WS2 layer, we anneal the 1T-WS2 sample at 200 °C (in the air for 30 min) for STEM imaging and find that the 1T phase is completely transformed into the 2H phase, as shown in Supplementary Figs. 8 and 9. Simultaneously, both the V atoms and S atoms are prominently discernible, in consistent with the STEM images of V-doped 2H-TMDs35,36,37. As a result, the substitutional V atoms are at an average concentration of 4.0 at% (~2.0 wt%) in the 1T-WS2 layer. To exclude the presence of V-based contaminations (e.g., V2O3, VO2, and VS2) in the transferred 1T sample, XPS spectra of 1T sample transferred on highly oriented pyrolytic graphite were performed (see details in Supplementary Figs. 10–12). The V signal in Supplementary Figs. 11c and 12c were ascribed to the V–S bond38, which was consistent with the EELS result in Fig. 2e.
The cross-sectional view STEM image of V SACs@1T-WS2 on the sapphire substrate was carried out to confirm the epitaxial relationship between V2O3 and the sapphire substrate. Figure 2g shows a low-resolution STEM image, indicating a ~20 nm thickness of V2O3 film. The inset high-resolution STEM image displays a sharp interface between V2O3 and sapphire substrate, revealing the epitaxial growth of V2O3 film on the sapphire substrate39. More importantly, we observe a single layer of WS2 from the sharp interface between V SACs@1T-WS2 and V2O3 (Fig. 2h). The lattice spacing of V2O3 measured in the STEM image is ~0.23 nm, in correspondence with the (006) plane of V2O3 obtained from the XRD diffraction pattern in Supplementary Fig. 239. In addition, the monolayer thickness of V SACs@1T-WS2 (before and after transferring) is also verified by atomic force microscope (AFM), as shown in Supplementary Fig. 13. Second-harmonic generation and angle-resolved-polarization Raman spectra (ARPRS) are commonly used to probe the symmetry and isotropic/anisotropic lattice structure of TMDs11,40,41. Therefore, the high symmetry and isotropic nature of V SACs@1T-WS2 have also been discussed in Supplementary Figs. 14 and 15.
Controlled phase conversion
The two different phases of 1T and 2H are easily highlighted by the confocal Raman spectroscopy, as provided in Fig. 3a, b. The Raman spectrum of V SACs@1T-WS2 exhibits two prominent peaks at 418.4 cm−1 and 354.6 cm−1 (Fig. 3a, orange plot), corresponding to the A1g and \({\mathrm{E}}^{1}_{{\mathrm{2g}}}\) resonance modes, respectively. Moreover, three additional typical peaks are observed at 147.9 cm−1 (J1), 214.5 cm−1 (J2), and 385.3 cm−1 (J3), respectively, which only exist in the metallic phase but not in 2H-WS2 monolayers (blue plot in Fig. 3a)1,16,18,22,42,43,44,45. Particularly, a prominently merged peak in the range of 150–250 cm−1 can be ascribed to hexagonal V2O3, as demonstrated in Supplementary Fig. 16. As verified in Supplementary Fig. 17, V2O3 film starts to nucleate when the temperature is elevated at ~600 °C. Hence, the different nucleation temperatures of V2O3 and WS2 give rise to the sequential growth of V2O3 and WS2 on the sapphire substrate. A brief discussion of the possible formation mechanism of the V2O3 film on the sapphire substrate is provided in Supplementary Fig. 17. In addition, we can see that the strongest photoluminescence (PL) signal (Fig. 3b, blue plot) is associated with monolayered 2H-WS231, while the negligible PL signal (Fig. 3b and Supplementary Fig. 18, orange plot) from V@ SACs@1T-WS2 is presumably due to the metallic nature1,2.

a Raman spectra of V2O3 film (black), V SACs@1T-WS2/V2O3 (orange), and 2H-WS2 (blue). b PL spectra of V2O3 film (black), V SACs@1T-WS2/V2O3 (orange), and 2H-WS2 (blue). c High-resolution XPS spectra of W 4f (left) and S 2p (right) core level peak regions for 2H and 1T phase, respectively. The fitting blue and pink curves represent the contributions of 1T and 2H phases, respectively. d Schematic representation of the 1T to 2H structural phase transition occurring upon thermal annealing. e Raman spectra of 2H-WS2, V SACs@1T-WS2, and V SACs@1T-WS2 with different annealing temperatures in H2/Ar for 2 h.
To illustrate the high purity of the obtained 1T phase, XPS spectra were performed to quantify the 1T and 2H compositions according to the high sensitivity of the tungsten signal to its oxidation state and coordination geometry1,46. The V@ SACs@1T-WS2 monolayers were transferred on the fresh sapphire substrate for the XPS measurements. The survey scan of V@ SACs@1T-WS2 was shown in Supplementary Fig. 19a. The O 1s signal in Supplementary Fig. 19b was ascribed to sapphire substrate. As provided in Fig. 3c, the W 4f core level peak of 1T phase is shifted to lower binding energy of ~1.0 eV than that of 2H phase, assigning to a major amount as high as 91% of 1T phase. The decline in binding energy is presumably caused by the chemical reduction of W from +4 to the +3 oxidation state47. The existence of 2H phase in the 1T sample may arise from the phase transition caused by the X-ray illumination, as no 2H characters have been detected from Raman spectra and STEM images, implying that a very high phase purity of the as-grown 1T sample (at least 91%). Notably, signals at higher binding energies of 36.3 eV and 38.5 eV from the 2H-WS2 stand for the peaks of W 5p3/2 and W6+ oxidation state species, respectively. Simultaneously, the high-resolution of core level S 2p peaks is coherently manifested lower binding energy in contrast to the S 2p peak from the 2H phase, consistent with the previous XPS studies of metallic WS21,22.
Moreover, the V SACs@1T-WS2 monolayers are extremely stable under ambient conditions even after one year, as shown in Supplementary Fig. 20. Importantly, a high 1T phase (~60%) is preserved in the sample from the XPS analysis in Supplementary Fig. 21b, and the decrease of 1T/2H ratio should be probably caused by the oxidation of V2O3 film on the surface, as demonstrated in Supplementary Fig. 21d. In addition, upon annealing at different temperatures (200 °C, 300 °C, and 400 °C, respectively) in H2/Ar condition, the 1T phase is partially transformed into a 2H phase. The schematic structure of phase transition from 1T to 2H is shown in Fig. 3d. From the XPS spectra recorded in Supplementary Fig. 22, despite gradual shrinkage of W 4f and S 2p peaks from 1T phase with the elevated temperature ≥300 °C, a very high amount of 1T phase is still preserved even after annealing at 400 °C (~49%). The different ratios of 1T/2H (W 4f) under different annealing temperatures were summarized in Supplementary Table 2. Raman spectra in Fig. 3e reveal that the enhanced intensities of \({\mathrm{E}}^{1}_{{\mathrm{2g}}}\)/A1g are associated with the decreasing 1T phase. Remarkably, characteristic peaks assigned to the 1T phase are still observed after annealing at 400 °C. Interestingly, as the increasing temperature, the PL intensity in Supplementary Fig. 23 was enhanced and largely blue-shifted, which was ascribed to the variation of the band-structure caused by the pronounced ratio of 2H phase in the 1T sample, in consistent with the previously reported metallic TMDs22.
Growth mechanism of V SACs@1T-WS2
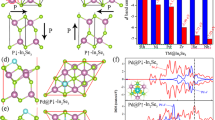
Our fabrication of V SACs@1T-WS2 monolayers by the one-step growth exemplifies the advantage of the CVD strategy over the exfoliated method and wet-chemical method, where the phase purity cannot be well controlled (Supplementary Table 1). The octahedral 1T phase has long been considered as an energetically unfavorable structure and tends to be transformed into a more stable 1T′ or 2H phase. Hence, understanding the growth mechanism of the present stable 1T structure is of importance for exploring more 1T-TMDs controllable growth. As demonstrated in Supplementary Fig. 17, the nucleation of V2O3 (at ~600 °C) precedes the epitaxial growth of WS2 (~800 °C) during the whole growth. Stepwise-products experiments were designed to explore the origin of the V SACs@1T-WS2 formation. The pristine one-step CVD growth was separated into two steps, that is initial V2O3 growth and subsequent V SACs@1T-WS2 growth, as depicted in Fig. 4a.

a Stepwise-products experiments for the V SACs@1T-WS2 growth process. b HRTEM image of V2O3-nuclei film formed in the upstream of the sapphire substrate. c High-resolution HAADF-STEM image of 1T-VS2 formed in the downstream of V2O3-nuclei film/sapphire substrate. The red spheres and yellow spheres represent the V atoms and S atoms, respectively, in the schematic model of 1T-VS2 in the basal plane. Inset: low-resolution of HAADF-STEM image of VS2 domain. d High-resolution HAADF-STEM image of V SACs@1T-WS2 formed on the surface of V2O3-nuclei film. The V atoms are highlighted by the dashed white circles. e The proposed growth mechanism of V SACs@1T-WS2 formed on the V2O3 film. f Computed binding energy of WS2 unit on 1T-WS2 vs 1T-VS2 on the surface of V2O3 (001). Scale bars: (b) 5 nm; (c) 0.5 nm, inset 50 nm; (d) 0.5 nm.
In the first growth step, VCl3 and sulfur were the co-precursors for the preparation of V2O3-nuclei film under the same growth condition with the pristine one-step growth. The presence of V2O3 was identified by the Raman spectrum in Supplementary Fig. 24a (blue plot) and XRD diffraction peaks in Supplementary Fig. 24b (blue plot). The oxygen element in the V2O3-nuclei film was presumably from the O2 residue in the tube furnace, as V2O3-No nuclei film was also synthesized if using VCl3 as the only precursor (Supplementary Fig. 24b, orange plot). Apart from the V2O3, trace amounts of VS2 were also identified based on the XPS spectra analysis (Supplementary Fig. 24d, f). To further confirm the presence of VS2, the V2O3-nuclei film was scraped from the sapphire substrate for the HRTEM measurement, as shown in Fig. 4b. In addition to the lattice spacing of 0.363 nm from V2O3, the other measured lattice spacing of ~0.286 nm can be well indexed with the (002) plane of 1T-VS2. Elemental mapping from the same region showed the uniform distributions of V, O, and S elements (Supplementary Fig. 25) in the V2O3-nuclei film, which was coincident with the analysis of the XPS survey spectrum in Supplementary Fig. 24c. More interestingly, 1T-VS2 nanocrystals were found on the surface of V2O3-nuclei film further downstream (Supplementary Fig. 26b, c, lower growth temperature zone, ~620 °C). In contrast to the trace amounts of VS2 nuclei found in the upstream (the hot center growth zone, ~860 °C), the VS2 nanocrystals downstream can be easily transferred from the V2O3-nuclei film using a standard transfer method for STEM imaging (Fig. 4c). The image showed a hexagonal atomic structure of 1T-VS2 in the [001] basal plane48, which was solid evidence that the VS2 intermediates were nucleated on the surface of V2O3 during the growth.
In the second step, the as-produced V2O3-nuclei film/sapphire substrate was put back into a fresh CVD process to collect the final product using WO3 and sulfur as the co-precursors. Intriguingly, it was found that smaller 1T domains of WS2 were formed on the surface of the film (Fig. 4d, Supplementary Fig. 27a, e), whereas 2H-WS2 domains were achieved if using V2O3-No nuclei film (Supplementary Fig. 27b, e, f) as the collecting substrate. Notably, nothing could be found if using commercial V2O3 (001) film (Supplementary Fig. 27c, d, and g), because that the rough surface and the low-quality of the commercial V2O3 were not in favor of the VS2 and WS2 nucleation on the surface. The 1T- or 2H-WS2 domains were also confirmed by the Raman mapping in Supplementary Figs. 28 and 29. As a result, these experimental observations imply that both the VS2 nucleation and the V2O3 film are very requisite for the V SACs@1T-WS2 growth and VS2 intermediates play the most important role in determining the 1T phase growth of WS2. The proposed growth mechanism is shown in Fig. 4e, in which the 1T-VS2 nuclei formed in the early growth stage serve as the 1T structure template and significantly strengthen the binding of WS2 unit (Fig. 4f, from 5.04 eV to 6.33 eV) on the V2O3 film, thereby triggering the epitaxial growth of the 1T phase nucleus. Such an epitaxially grown WS2 layer should be the 1T phase instead of the 2H phase due to the much higher binding energy of 1T-VS2/1T-WS2 (6.09 eV) than 1T-VS2/2H-WS2 (4.19 eV) (Supplementary Fig. 30). The computed lattice parameters of 2H-WS2, V SACs@2H-WS2, and V SACs@1T-WS2 are shown in Supplementary Table 3. In addition, it was also demonstrated that the amount of VCl3 could significantly affect the controllable phase growth of WS2 (see details in Supplementary Figs. 31–34). The influence of heating temperature on the synthesis of 1T-WS2 has also been investigated, as displayed in Supplementary Figs. 35–37. Moreover, vanadocene precursors were also investigated to enrich the growth method of 1T-WS2 monolayers (Supplementary Fig. 38).
HER activity of V SACs@1T-WS2
The as-produced V SACs@1T-WS2 monolayers were transferred on the glass carbon (GC) electrode for the HER performance measurement using a three-electrode setup in 0.5 M H2SO4. In addition, HER performance of the other investigated electrocatalysts, i.e., bare GC, 2H-WS2, 1T-400 (V SACs@1T-WS2 annealed at 400 °C in H2/Ar for 2 h), 2H1T (transformed by V SACs@1T-WS2 annealed at 200 °C in the air for 30 min) and commercial Pt/C-20 % were evaluated for comparison. As shown in linear sweep voltammetry (LSV) curves (Fig. 5a), V SACs@1T-WS2 exhibited a low overpotential of 185 mV (η10) at a current density of 10 mA/cm2 with an ultralow mass loading of 1.8–6.5 μg/cm2 (see details in Supplementary Figs. S39–S45 and Supplementary Table 4–6), outperforming the 1T-400 (blue plot, η10 = 325 mV), 2H1T (olive plot, η10 = 515 mV) and 2H (red plot, η10 = 684 mV) electrodes. The excellent activity of SACs@1T-WS2 monolayers was further demonstrated by the comparisons of Tafel slopes for different catalysts, as shown in Fig. 5b. The Tafel slope for V SACs@1T-WS2 was 61 mV/dec (Fig. 5c), which is much lower than that of 1T-400 (100 mV/dec), 2H1T (103 mV/dec), and 2H-WS2 (194 mV/dec). Please note that mass loading of electrocatalysts has a great impact on the measured activities; hence, it has generally used as catalyst performance metrics7. Although the mass loading of SACs@1T-WS2 was much lower than most of the non-precious electrocatalysts (Supplementary Fig. 46), the HER performance was still comparable to that of strained 1T′-WS2 nanosheets (NSs)1, 1T′-MoS2 NSs2, 1T′-MoS2 monolayers11, and better than that of 2H-1T boundaries MoS2 monolayers49, 1T′ WSe2 NSs7 and PE-CVD 1T-WS2 film15. The electrochemical surface area (ECSA) was calculated as an important factor to affect the catalytic activity of electrocatalysts50,51,52. The calculated details were shown in the experimental section and Supplementary Fig. 47. Double-layer capacitance (Cdl) and ECSA values were 139.5 μF/cm2 and 3.49 cm2 for V SACs@1T-WS2 and 61.7 μF/cm2 and 1.54 cm2 for 2H-WS2, suggesting the critical contributions of V atom sites and high purity of 1T-WS2. The TOF values of the SACs@1T-WS2 (Fig. 5c) were obtained according to the precious reports23,53 and calculated to be 3.01 s−1, 12.78 s−1, and 24.15 s−1 at overpotentials of 100 mV, 150 mV, and 200 mV, respectively, which were much higher than those recently representative electrocatalysts listed in Fig. 5d and Supplementary Table 7.

a LSV curves of GC, 2H (2H-WS2), 1T-400 (V SACs@1T-WS2 annealed at 400 °C in H2/Ar for 2 h), 2H1T (V SACs@2H-WS2, transformed by V SACs@1T-WS2), Pt/C (Pt/C 20%) and V SACs@1T-WS2 electrodes in 0.5 M H2SO4 with a scan rate of 5 mV/s. b Tafel plots of 2H, 1T-400, 2H1T, Pt/C 20% and V SACs@1T electrodes. c TOF values of V SACs@1T-WS2 measured at different overpotentials. d TOF comparison with other representative electrocatalysts. e HER activity as a function of the 1T phase ratios obtained by annealing V SACs@1T-WS2 in the H2/Ar for 2 h. f Electrochemical stability of V SACs@1T-WS2 after 2000 cycles of CV tests.
As the V SACs@1T-WS2 monolayers contain a high concentration of 1T phase, to investigate the influence of 1T content on the catalytic performance, the V SACs@1T-WS2 electrode was gradually annealed at different temperatures and the HER activity after each annealing treatment was performed, ensuring that the identical dimensions and geometrical areas to evaluate the catalytic properties. Remarkably, the HER activities were strongly related to the 1T phase content as demonstrated by a gradual decrease in the η10 with decreasing 1T ratios (Fig. 5e). Remarkably, negligible activity degradation can be observed in the reproducible polarization curve of V SACs@1T-WS2 in Fig. 5f even after 2000 continuous cycling, indicative of a good HER performance stability. The stability test was conducted at current densities higher than 10 mA/cm2 in 0.5 M H2SO4 electrolyte for 100 h. As revealed by the chronoamperometric curve of V SACs 1T-WS2 electrocatalyst in Supplementary Fig. 48, the current density for the V SACs 1T-WS2 electrocatalyst displayed a slight current decay of 1.0 mA cm−2 after 24 h and 3.4 mA cm−2 after 100 h, indicating high stability of V SACs 1T-WS2 catalyst. The metallic properties of V SACs 1T-WS2 catalyst after stability test were also investigated by Raman spectroscopy (Supplementary Fig. 49a), which showed obvious metallic peaks (J1, J2, J3) in the Raman spectrum (red plot). Moreover, the STEM image showed in Supplementary Fig. 49b confirmed the V SACs 1T-WS2 structure after the stability test. Both the Raman spectrum and STEM image indicated the robust 1T structure of V SACs 1T-WS2 catalyst after HER test.
V SACs enhancing the HER activity of 1T-WS2
Especially, the HER performance of 2H-WS2, V SACs@2H-WS2, and V SACs@1T-WS2 was further studied using DFT considering both basal plane (Supplementary Fig. 50) and edge sites (Supplementary Figs. 51–53) as the active sites. Our calculations show that the V SACs could significantly influence the free energy of H adsorption (∆GH) on the edge sites of 1T-WS2, as summarized in Supplementary Table 8. Please note that the |∆GH| of V SACs@1T-WS2 at the basal plane is 0.4 eV, which is approximate to the |∆GH| of intrinsic 1T-WS2 at 0.28 eV1, implying that the single-atom V sites have a negligible influence on the HER performance of 1T-WS2 in the basal plane. Compared to the 2H-WS2 and V SACs@2H-WS2, V SACs@1T-WS2 exhibits the lowest |∆GH| (0.05 eV) at V-atom sites (Fig. 6a), indicating that the isolated V atoms are catalytically active in the layer of 1T-WS2. The charge depletion at the active sites has been proven to play an essential role in improving the electrochemical activity of the catalysts13,54. To acquire a deeper understanding of how single V atoms enhanced the activity of 1T-WS2, the charge redistribution of V SACs@1T-WS2 was studied. As shown in the inset image in Fig. 6b, we can clearly see that when one W atom was substituted by a V atom, there was more charge depletion generated at the V-atom site. Such variations in the local electronic structure can also be well described using the d-band theory55. A linear inverse correlation between |∆GH| and the d-band center at the most active edge sites were revealed, indicating that a more negative d-band center corresponds to more occupation of the antibonding states, resulting in weaker H adsorption of V SACs@1T-WS2 comparable to their 2H counterparts.

a The Gibbs free energy of H adsorption (ΔGH*) of 2H-WS2, V SACs@2H-WS2, and V SACs@1T-WS2 on the V and S edge sites. b Relationship between |∆GH*| and the d-band center of adjacent W/V ions at the most active edge sites of 2H-WS2, V SACs@2H-WS2, and V SACs@1T-WS2.
Discussion
In summary, we have demonstrated a direct synthesis of single-atom V sites on the high purity 1T-WS2 monolayers via a one-step CVD strategy through introducing VCl3 as a co-precursor, with much better HER performance than the 2H counterparts. The step-wise experimental findings together with DFT results shed light on the understanding of the CVD-grown V SACs@1T-WS2 monolayers, that is the 1T-VS2 nuclei initially act as the 1T template structures for the follow-up V SACs@1T-WS2 growth on the V2O3 film. The HER performance of intrinsic 1T-WS2 was remarkably boosted after the modification of isolated V atoms. DFT calculations highlight that the highly activated V-atom sites primarily account for the excellent HER activity of V SACs@1T-WS2 monolayers. Our findings could fill the gap of SACs grown via a one-step CVD strategy and pave the way to design more efficient and robust electrocatalysts for applications in water splitting.
Methods
The growth of V SACs@1T-WS2 monolayers, 2H-WS2 monolayers, V2O3 film
In the growth of monolayer V SACs@1T-WS2, sulfur (Sigma-Aldrich, 99%), tungsten trioxide (WO3, Sigma-Aldrich, 99.99%), and vanadium (III) chloride (VCl3, Sigma-Aldrich, 99%,) were used as the growth precursors. Two quartz boats with 0.03 g of VCl3 powder (in the hot center) and 0.30 g WO3 powder (the upstream of VCl3) were placed sequentially in the hot center of the furnace. And a fresh sapphire (c-plane) substrate (1 cm × 2 cm) was placed face-down of the quartz boat to collect the final product. The heating temperature was kept at 860 °C. The S powder was placed in a separate quartz boat at the upper stream side of the cold zone and the temperature was maintained at 160 °C during the reaction. The gas flow has consisted of an ultrapure mixed Ar/H2 (Ar = 80 sccm, H2 = 20 sccm), and the chamber pressure was controlled at 10 Torr. After reaching the desired growth temperature of 860 °C, the heating zone was kept for 15 min and the furnace was then naturally cooled down to room temperature. The V2O3 film was simultaneously formed during the V SACs@1T-WS2 growth. The commercial V2O3 (001) film on the c-plane sapphire was purchased from Xi’an Qiyue Biotechnology Co., Ltd. To obtained 2H-WS2 monolayers, we used WO3 and suffer as the precursors with the same growth condition with V SACs@1T-WS2. To obtain V2O3-No nuclei film, we used the only VCl3 as the precursors with the same growth condition. To obtain V2O3-nuclei film, we used only VCl3 and sulfur as the precursors with the same growth condition.
V2O3 film growth mechanism and characterization
According to the lattice parameters of V2O3 and sapphire (V2O3: hexagonal, a = 0.492 nm, c = 1.397 nm; α-Al2O3: hexagonal, a = 0.475 nm, c = 1.297 nm), hexagonal V2O3 shares the same lattice-type and similar lattice parameters with α-Al2O3; thus, it is expected to epitaxially grow on α-Al2O3 with the relationship V2O3 (001)[100]//α-Al2O3 (001)[100]39,56,57,58.
To investigate the growth mechanism of V2O3 film, we prepared V2O3-No nuclei film under different temperatures, as exhibited in Supplementary Fig. 17a. It can be observed that V2O3 film could be formed at ≥600 °C during the temperature-dependent controllable experiments. Particularly, a merged peak in the range of 150–250 cm−1 was observed in the Raman spectrum and the peak was correlated to both monoclinic A1g and hexagonal V2O3 A1g symmetry, indicative of a mixed phase. However, the additional peaks of low intensity at 300 cm−1 and 500 cm−1 were solely attributable to hexagonal V2O359,60, indicating the hexagonal structure of the as-grown V2O3 film. Under the temperature of 600 °C, low-quality film or nothing will be formed on the sapphire substrate. Simultaneously, we measured the Raman spectra of the powders from the VCl3 quarts boat after each growth reaction with different growth temperatures, as shown in Supplementary Fig. 17b. VO2 (B) will be evidently formed after the decomposition of VCl361 (see Eqs. (1) and (2) in Supplementary Fig. 17).
Transfer process of V SACs@1T-WS2, 2H-WS2, V2O3-nuclei film
The as-grown samples were transferred onto arbitrary substrates, such as fresh sapphire, SiO2/Si, GC, and holy-carbon nickel TEM Grid using a modified method in our lab. First, poly(methyl methacrylate) (PMMA) was spin-coated onto the samples followed by 120 °C baking for 10 min, and then etched by 5% HF solution for 2–3 min. The PMMA/sample was gently peeled off by the tweezers and deposited onto the targeted substrates. PMMA/sample/targeted substrate was baked for 1 h at 120 °C in air. The PMMA was removed by acetone and cleaned with isopropyl alcohol. The V2O3-nuclei film was scratched by tweezers and ultrasonic the V2O3/sapphire sample in the ethanol solution for 15 min for the HRTEM measurement. The sample was prepared by dropping the solution onto the holy-carbon nickel TEM grid.
For the STEM measurements samples, the extra annealing process was necessary to remove the residual PMMA. For the 2H-WS2 sample annealing, the sample was located in a vacuum container (less than 10−6 Torr) at 350 °C overnight. For the V SACs@1T-WS2 sample annealing, the sample was located in a vacuum container (less than 10−6 Torr) at 200 °C for 2 h. For the XPS and Raman spectra of V SACs@1T-WS2 annealed at different temperatures, the V SACs@1T-WS2 monolayers were transferred on the fresh sapphire substrates and annealed in H2/Ar condition with different temperatures.
Characterization
Optical spectroscopy is collected under a Witec alpha 300 R confocal Raman microscopic system. Gratings of 1800 lg/mm and 300 lg/mm are selected for the high-resolution Raman spectrum and wide range PL spectrum, respectively. The TMDs are excited by 532 nm laser with a power of 1 mW and spot size of 0.5 μm and emitted Raman signal is collected by 100× objective (N.A = 0.9) from a Carl Zeiss Microscopy. ARPRS are conducted on a triple-grating micro-Raman spectrometer (Horiba-JY T64000) with a 532 nm laser under a backscattering configuration. The polarization is resolved by rotating sample orientation on the normal axis of the basal plane. The emitted Raman signal is collected through a 100× objective, dispersed with 1800 lg/mm grating, and detected by a charge-coupled device.
An AFM (Cypher ES environmental AFM) was utilized to obtain the morphology images and the height profile of V SACs@WS2 monolayers. The crystalline of the different samples was probed using a Bruker D8 advance powder XRD with Cu Ka radiation. X-ray photoelectron spectroscopy (XPS) studies were carried out in a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) under a vacuum of 1 × 10−9 mbar. The spectra were collected at fixed analyzer pass energies of 160 eV and 20 eV. The binding energies in XPS analysis were corrected by referencing C 1s line at 284.8 eV. STEM and TEM images were performed by Titan 40-300 Themis Z TEM from Thermo Fisher, USA (former FEI Co) equipped with a double Cs corrector, an electron monochromator, and a Gatan imaging filter quantum 966. The microscope was operated at 80 kV to minimize electron beam induced. Probe semi convergence angle was tuned to 30 mrad and probe current to 50 pA. For high-angle annular dark-field (HAADF) STEM images the inner collection angle was about 80 mrad. Radial wiener filter was carried out to enhance the visibility of atoms
Computational details
First-principles simulations were performed at the DFT level implemented by the Vienna ab initio simulation package62,63,64,65 with the all-electron projector augmented wave model66 and Perdew–Burke–Ernzerhof functions67. The DFT-D3 method was applied to include vdW interaction corrections68. A kinetic energy cutoff of 400 eV was used for the plane-wave expansion of the electronic wave function. The convergence criteria of force and energy were set as 0.01 eVÅ−1 and 10−5 eV, respectively. Gaussian smearing of 0.1 eV was applied for optimization. A k-point grid with a 4 × 4 × 1 gamma-centered mesh was used for the WS2 unit cell. For supercells that contain a larger number of vanadium and oxygen atoms, a corresponding number of k-points were used to keep the k-mesh spacing constant across different structures. The climbing image nudged elastic band was applied for transition state optimization69.
The free energy of the adsorption atomic hydrogen (ΔGH) is obtained by ΔGH = ΔEH + ΔEZPE − TΔSH. ΔEH is the adsorption energy defined by ΔEH = Esur − H − Esur + ½ EH2. According to previous reports, ΔGH can be written as ΔGH = ΔEH + 0.25, where 0.25 eV is the contribution from ZPE and entropy at 298 K70.
Electrochemical measurements
Electrochemical measurements were carried in a PGSTAT 302N Autolab Potentiostat/Galvanostat (Metrohm) at room temperature. Graphite rod and Ag/AgCl (in a saturated KCl solution) electrodes were employed as the counter and reference electrodes, respectively. The V SACs@1T-WS2 and 2H-WS2 were transferred onto the GC electrodes as the working electrodes and dried. Nafion solution (0.5%) was drop-cast to protect WS2 film. The V SACs@1T-400 electrode was prepared by annealing the V SACs@1T-WS2 electrode at 400 °C in H2/Ar for 2 h. The 2H1T electrode was prepared by annealing the V SACs@1T-WS2 electrode at 200 °C in the air for 30 min. The HER activities of different samples were evaluated by measuring polarization curves with LSV at a scan rate of 0.5 mV/s in 0.5 M H2SO4 solutions. Potentials were referenced to a reversible hydrogen electrode (RHE). The commercial Pt/C (20 wt% Pt on Vulcan carbon black) supported by GC was prepared by mixing the Pt/C, nafion, and isopropanol, sonicating for 30 min, and drop-casting on the GC. The mass loading of Pt/C was 500 μg/cm2. The potential cycling was performed between 0.197 and −0.6 V vs RHE at 5 mV s−1. All data have been corrected for a small ohmic drop based on impedance spectroscopy. ECSA and Cdl are determined by cyclic voltammograms at various scan rates (10, 30, 50, 70, 90, 110. 130, 150, 170, 190, and 210 mV/s) in the potential range (0.15–0.35 V vs. RHE). The capacitive currents (ΔJ) are plotted as a function of scan rate and Cdl is equal to half of the slope. The reference specific capacitance (Cs) of 40 μF/cm2 is used in this work. The ECSA for the different catalysts are achieved based on the following equation
Calculation of turnover frequency
The TOF calculation details were specified as below, which was reported elsewhere23,53.
The total hydrogen turnovers were calculated from the current density in the LSV polarization curve according to the equation as below:
The number of active sites in the V SACs@1T-WS2 catalyst was obtained from the mass loading on the glass carbon electrode.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Voiry, D. et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 12, 850–855 (2013).
Yu, Y. et al. High phase-purity 1T′-MoS2- and 1T′-MoSe2-layered crystals. Nat. Chem. 10, 638–643 (2018).
Liao, G. et al. Semiconductor polymeric graphitic carbon nitride photocatalysts: the “holy grail” for the photocatalytic hydrogen evolution reaction under visible light. Energy Environ. Sci. 12, 2080–2147 (2019).
Liao, G. et al. Ag-based nanocomposites: synthesis and applications in catalysis. Nanoscale 11, 7062–7096 (2019).
Thalluri, S. M. et al. Inverted pyramid textured p-silicon covered with Co2P as an efficient and stable solar hydrogen evolution photocathode. ACS Energy Lett. 4, 1755–1762 (2019).
Chen, X. et al. High phase-purity 1T-MoS2 ultrathin nanosheets by a spatially confined template. Angrew. Chem. Int. Ed. 58, 17621 (2019).
Sokolikova, M. S., Sherrell, P. C., Palczynski, P., Bemmer, V. L. & Mattevi, C. Direct solution-phase synthesis of 1T′ WSe2 nanosheets. Nat. Commun. 10, 712 (2019).
Kuznetsov, D. A. et al. Single site cobalt substitution in 2D molybdenum carbide (MXene) enhances catalytic activity in the hydrogen evolution reaction. J. Am. Chem. Soc. 141, 17809–17816 (2019).
Fu, W. et al. Epitaxial growth of graphene on V8C7 nanomeshs for highly efficient and stable hydrogen evolution reaction. J. Catal. 369, 47–53 (2019).
Jaramillo, T. F. et al. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 317, 100–102 (2007).
Liu, L. et al. Phase-selective synthesis of 1T′ MoS2 monolayers and heterophase bilayers. Nat. Mater. 17, 1108–1114 (2018).
Duerloo, K.-A. N., Li, Y. & Reed, E. J. Structural phase transitions in two-dimensional Mo-and W-dichalcogenide monolayers. Nat. Commun. 5, 4214 (2014).
Deng, S. et al. Synergistic doping and intercalation: realizing deep phase modulation on MoS2 arrays for high-efficiency hydrogen evolution reaction. Angew. Chem. Int. Ed. 58, 16289–16296 (2019).
Enyashin, A. N. et al. New route for stabilization of 1T-WS2 and MoS2 phases. J. Phys. Chem. C 115, 24586–24591 (2011).
Kim, H.-U. et al. Wafer-scale and low-temperature growth of 1T-WS2 film for efficient and stable hydrogen evolution reaction. Small 16, 1905000 (2020).
Liu, Q. et al. Stable metallic 1T-WS2 nanoribbons intercalated with ammonia Ions: the correlation between structure and electrical/optical properties. Adv. Mater. 27, 4837–4844 (2015).
Lukowski, M. A. et al. Enhanced hydrogen evolution catalysis from chemically exfoliated metallic MoS2 nanosheets. J. Am. Chem. Soc. 135, 10274–10277 (2013).
Mahler, B., Hoepfner, V., Liao, K. & Ozin, G. A. Colloidal synthesis of 1T-WS2 and 2H-WS2 nanosheets: applications for photocatalytic hydrogen evolution. J. Am. Chem. Soc. 136, 14121–14127 (2014).
Park, J. C. et al. Phase-engineered synthesis of centimeter-scale 1T′- and 2H-molybdenum ditelluride thin films. ACS Nano 9, 6548–6554 (2015).
Voiry, D. et al. Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction. Nano Lett. 13, 6222–6227 (2013).
Yin, Y. et al. Contributions of phase, sulfur vacancies, and edges to the hydrogen evolution reaction catalytic activity of porous molybdenum disulfide nanosheets. J. Am. Chem. Soc. 138, 7965–7972 (2016).
Zhao, X., Ma, X., Sun, J., Li, D. & Yang, X. Enhanced catalytic activities of surfactant-assisted exfoliated WS2 nanodots for hydrogen evolution. ACS Nano 10, 2159–2166 (2016).
Qi, K. et al. Single-atom cobalt array bound to distorted 1T MoS2 with ensemble effect for hydrogen evolution catalysis. Nat. Commun. 10, 5231 (2019).
Zhang, H., Yu, L., Chen, T., Zhou, W. & Lou, X. W. Surface modulation of hierarchical MoS2 nanosheets by Ni single atoms for enhanced electrocatalytic hydrogen evolution. Adv. Funct. Mater. 28, 1807086 (2018).
Deng, J. et al. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 8, 1594–1601 (2015).
Chow, P. K. et al. Wetting of mono and few-layered WS2 and MoS2 films supported on Si/SiO2 substrates. ACS Nano 9, 3023–3031 (2015).
Elías, A. L. et al. Controlled synthesis and transfer of large-area WS2 sheets: from single layer to few layers. ACS Nano 7, 5235–5242 (2013).
Gutiérrez, H. R. et al. Extraordinary room-temperature photoluminescence in triangular WS2 monolayers. Nano Lett. 13, 3447–3454 (2013).
Perea-López, N. et al. Photosensor device based on few-layered WS2 films. Adv. Funct. Mater. 23, 5511–5517 (2013).
Cui, Y. et al. High-performance monolayer WS2 field-effect transistors on high-k dielectrics. Adv. Mater. 27, 5230–5234 (2015).
Gao, Y. et al. Large-area synthesis of high-quality and uniform monolayer WS2 on reusable Au foils. Nat. Commun. 6, 8569 (2015).
Yang, W. et al. Electrically tunable valley-light emitting diode (vLED) based on CVD-grown monolayer WS2. Nano Lett. 16, 1560–1567 (2016).
Jeong, H. Y. et al. Heterogeneous defect domains in single-crystalline hexagonal WS2. Adv. Mater. 29, 1605043 (2017).
Laffont, L. et al. High resolution EELS of Cu–V oxides: application to batteries materials. Micron 37, 459–464 (2006).
Zhang, F. et al. Monolayer vanadium-doped tungsten disulfide: a room-temperature dilute magnetic semiconductor. Adv. Sci. 7, 2001174 (2020).
Pham, Y. T. H. et al. Tunable ferromagnetism and thermally induced spin flip in vanadium-doped tungsten diselenide monolayers at room temperature. Adv. Mater. 32, 2003607 (2020).
Yun, S. J. et al. Ferromagnetic order at room temperature in monolayer WSe2 semiconductor via vanadium dopant. Adv. Sci. 7, 1903076 (2020).
Yu, S. H. et al. In situ hybridizing MoS2 microflowers on VS2 microflakes in a one-pot CVD process for electrolytic hydrogen evolution reaction. ACS Appl. Energy Mater. 2, 5799–5808 (2019).
Sun, G., Cao, X., Long, S., Li, R. & Jin, P. Optical and electrical performance of thermochromic V2O3 thin film fabricated by magnetron sputtering. Appl. Phys. Lett. 111, 053901 (2017).
Beams, R. et al. Characterization of few-layer 1T′ MoTe2 by polarization-resolved second harmonic generation and Raman scattering. ACS Nano 10, 9626–9636 (2016).
Wang, Y. et al. Structural phase transition in monolayer MoTe2 driven by electrostatic doping. Nature 550, 487 (2017).
Loh, T. A. & Chua, D. H. Origin of hybrid 1T-and 2H-WS2 ultrathin layers by pulsed laser deposition. J. Phys. Chem. 119, 27496–27504 (2015).
Loh, T. A. J., Chua, D. H. C. & Wee, A. T. S. One-step synthesis of few-layer WS2 by pulsed laser deposition. Sci. Rep. 5, 18116 (2015).
Tan, S. J. et al. Chemical stabilization of 1T′ phase transition metal dichalcogenides with giant optical Kerr nonlinearity. J. Am. Chem. Soc. 139, 2504–2511 (2017).
Tsai, H.-L., Heising, J., Schindler, J. L., Kannewurf, C. R. & Kanatzidis, M. G. Exfoliated−restacked phase of WS2. Chem. Mater. 9, 879–882 (1997).
Chen, W. et al. Quantum dots of 1T phase transitional metal dichalcogenides generated via electrochemical Li intercalation. ACS Nano 12, 308–316 (2018).
Kappera, R. et al. Phase-engineered low-resistance contacts for ultrathin MoS2 transistors. Nat. Mater. 13, 1128 (2014).
Yuan, J. et al. Facile synthesis of single crystal vanadium disulfide nanosheets by chemical vapor deposition for efficient hydrogen evolution reaction. Adv. Mater. 27, 5605–5609 (2015).
Zhu, J. et al. Boundary activated hydrogen evolution reaction on monolayer MoS2. Nat. Commun. 10, 1348 (2019).
Anjum, M. A. R., Jeong, H. Y., Lee, M. H., Shin, H. S. & Lee, J. S. Efficient hydrogen evolution reaction catalysis in alkaline media by all-in-one MoS2 with multifunctional active sites. Adv. Mater. 30, 1707105 (2018).
McCrory, C. C. L. et al. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 137, 4347–4357 (2015).
Voiry, D. et al. Best practices for reporting electrocatalytic performance of nanomaterials. ACS Nano 12, 9635–9638 (2018).
Wan, J. et al. In situ phosphatizing of triphenylphosphine encapsulated within metal–organic frameworks to design atomic Co1-P1N3 interfacial structure for promoting catalytic performance. J. Am. Chem. Soc. 142, 8431–8439 (2020).
Zhao, Z., Li, M., Zhang, L., Dai, L. & Xia, Z. Design principles for heteroatom-doped carbon nanomaterials as highly efficient catalysts for fuel cells and metal-air batteries. Adv. Mater. 27, 6834–6840 (2015).
Chen, H. et al. Promoting subordinate, efficient ruthenium sites with interstitial silicon for Pt-Like electrocatalytic activity. Angew. Chem. Int. Ed. 58, 11409–11413 (2019).
Ji, Y. D. et al. Epitaxial growth and metal-insulator transition of vanadium oxide thin films with controllable phases. Appl. Phys. Lett. 101, 071902 (2012).
Allimi, B. S. et al. Growth of V2O3 thin films on a-plane (110) and c-plane (001) sapphire via pulsed-laser deposition. J. Mater. Res. 22, 2825–2831 (2007).
Sakai, J., Limelette, P. & Funakubo, H. Transport properties and c/a ratio of V2O3 thin films grown on C- and R- plane sapphire substrates by pulsed laser deposition. Appl. Phys. Lett. 107, 241901 (2015).
Kafizas, A., Hyett, G. & Parkin, I. P. Combinatorial atmospheric pressure chemical vapour deposition (cAPCVD) of a mixed vanadium oxide and vanadium oxynitride thin film. J. Mater. Chem. 19, 1399–1408 (2009).
Kuroda, N. & Fan, H. Y. Raman scattering and phase transitions of V2O3. Phys. Rev. B 16, 5003–5008 (1977).
Xu, H. F., Liu, Y., Wei, N. & Jin, S. W. From VO2(B) to VO2(A) nanorods: hydrothermal synthesis, evolution and optical properties in V2O5H2C2O4H2O system. Optik 125, 6078–6081 (2014).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. B 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 132, 154104 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Tang, Q. & Jiang, D.-E Mechanism of hydrogen evolution reaction on 1T-MoS2 from first principles. ACS Catal. 6, 4953–4961 (2016).
Acknowledgements
L.L. acknowledges support from King Abdullah University of Science and Technology (Saudi Arabia). Y.L. and D.W. acknowledge support from the National Key R&D Program of China (2018YFA0702003) and the National Natural Science Foundation of China (21890383, 21671117, 21871159). W.Z. acknowledges support from the Educational Commission of Guangdong Province project (No.2015KGJHZ006).
Author information
Authors and Affiliations
Contributions
Y.L., L.L., and D.W. conceived the idea and designed the research project. A.H. designed the synthesis and performance experiments, collected and analyzed the data, and wrote the manuscript. X.Z. contributed to the characterizations of samples and wrote the manuscript. X.W. contributed to the computational results and wrote the manuscript. S.L. designed the optical characterizations experiments, analyzed the data, and wrote the manuscript. L.G. and Q.Z. designed the simulation STEM analyzes. Q.X., Z.Z., F.L., W.Z. contributed to revising the manuscript. All the authors commented on the manuscript and have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Baizeng Fang, Shaojun Guo, and other, anonymous reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, A., Zhou, X., Wang, X. et al. One-step synthesis of single-site vanadium substitution in 1T-WS2 monolayers for enhanced hydrogen evolution catalysis. Nat Commun 12, 709 (2021). https://doi.org/10.1038/s41467-021-20951-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-20951-9
This article is cited by
-
Regulating the conductance of tungsten diselenide by oxygen plasma and improving its electrical stability by encapsulation
Nano Research (2024)
-
Diverse atomic structure configurations of metal-doped transition metal dichalcogenides for enhancing hydrogen evolution
Nano Research (2024)
-
Designing multi-heterogeneous interfaces of Ni-MoS2@NiS2@Ni3S2 hybrid for hydrogen evolution
Nano Research (2024)
-
Electric double layer-mediated polarization field for optimizing photogenerated carrier dynamics and thermodynamics
Nature Communications (2023)
-
Controllable substitutional vanadium doping in wafer-scale molybdenum disulfide films
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
