
Abstract
Elucidating atmospheric oxidation mechanisms is necessary for estimating the lifetimes of atmospheric species and understanding secondary organic aerosol formation and atmospheric oxidation capacity. We report an unexpectedly fast mechanistic pathway for the unimolecular reactions of large stabilized Criegee intermediates, which involves the formation of bicyclic structures from large Criegee intermediates containing an aldehyde group. The barrier heights of the mechanistic pathways are unexpectedly low – about 2–3 kcal/mol – and are at least 10 kcal/mol lower than those of hydrogen shift processes in large syn Criegee intermediates; and the calculated rate constants show that the mechanistic pathways are 105-109 times faster than those of the corresponding hydrogen shift processes. The present findings indicate that analogous low-energy pathways can now also be expected in other large Criegee intermediates and that oxidative capacity of some Criegee intermediates is smaller than would be predicted by existing models.
Similar content being viewed by others
Introduction
Ozonolysis of alkenes is an important step in atmospheric mechanisms that control the oxidative capacity of the atmosphere and the production of sulfuric acid and secondary organic aerosols that are important in climate models1,2,3,4,5.
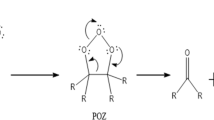
The first step in ozonolysis is 1,3-dipolar cycloaddition of O3 to the alkene to produce a primary ozonide (POZ), which is a compound containing an O–O–O group. The POZs produced by ozonolysis of acyclic or exocyclic alkenes decompose to produce a carbonyl oxide—called a Criegee intermediate (CI)—and an aldehyde or ketone. The reaction is exothermic, and as a consequence the CI is produced hot6, which is called chemical activation. Previous investigation has shown that hot syn CIs from cycloalkene ozonolysis mainly undergo a hydrogen shift reaction to produce a vinyl hydroperoxide6, which decomposes to produce a hydroxyl radical; this mechanism is called the prompt OH mechanism. But a fraction of the CIs are collisionally thermalized, producing what are called stabilized CIs or SCIs. The SCIs may also undergo hydrogen shift reactions, but their longer life allows other reactions to compete, especially unimolecular rearrangement to dioxiranes and bimolecular reactions with a variety of atmospheric constituents, such as water, water dimers, SO2, or other acids6,7,8,9,10,11,12,13.
The POZs produced by ozonolysis of endocyclic alkenes produce bifunctional molecules containing both a carbonyl oxide group and a carbonyl group. They may also react by a hydrogen shift, a unimolecular rearrangement to produce a dioxirane, or by a bimolecular reaction, but they also have other possibilities such as intramolecular attack of the carbonyl oxide on the carbonyl, producing a secondary ozonide (SOZ), which is a compound containing an O–C–O–O group.
Direct detection of gaseous Criegee intermediates is mainly limited to small CIs8,9,10,11,12,13,14,15,16. Larger CIs capable of producing SOZs include those from ozonolysis of cyclic terpenes and terpenoids, which are volatile organic compounds with high biogenic concentrations in the atmosphere. Experimental data on the large Criegee intermediates are scarce. Here, we investigate the unimolecular reactions two large Criegee intermediates, namely C5H8O3 and C6H10O3, which have structural features that may be considered as prototypes for CIs produced by ozonolysis of cyclic terpenes. The hydrogen shift and dioxirane cyclization reactions are shown in Fig. 1, in which the reactions labeled Z are for syn species and those labeled E are for anti.

Hydrogen shift and dioxirane cyclization reactions
In the present paper, we study reactions of SCIs, and we report reactions that in the syn case are much faster than the above hydrogen shift reactions that are used in standard mechanisms. In particular, we report that the bicyclic ring closure reactions as shown in Fig. 2 are much faster than the hydrogen shift for the syn case and of comparable reaction rate to the dioxirane cyclization reaction for the anti case. We show here that these reactions are important when the Criegee intermediates have aldehyde functional groups, but they should probably also be important when they have other functional groups such as carboxylic acid functional groups. For example, monoterpenes17 such as limonene, 2-carene, 3-carene, phellandrene, terpinene, and terpinolene have cyclic structures whose ozonolyisis leads to the formation of CHO and HCOO groups. The Criegee intermediates resulting from ozonolysis of unsaturated oxygenated organic compounds18 such as unsaturated aldehydes19 and fatty acids20 may also contain other groups.

Bicyclic ring closure reactions
Results
General remarks
The importance of including multiple torsions and multiple transition states in reactions of large molecules has been emphasized previously21,22. In the present work we employ multistructural variational transition state theory22 to include all rotamer structures in our rate calculations; further details of the kinetics methods are given in the Methods section.
Although we use standard notation, it is worthwhile to review some definitions since some readers may not know them. The classical barrier height is the difference in Born–Oppenheimer potential energy between the conventional transition structure (saddle point) and the equilibrium reactant. When we give enthalpies of activation in this article, they are the values calculated by conventional transition state theory in which the transition state is located at the saddle point, and they are for 0 K; thus we label these quantities at \(\Delta H_0^\ddagger\), and they are equal to the classical barrier height plus the zero-point energy of the transition structure minus the zero-point energy of the reactant. Activation energies are temperature-dependent and are defined in terms of Arrhenius plots as −Rdlnk/d(1/T), where R is the gas constant, k is the rate constant, and T is the temperature.
Large Criegee intermediates have many conformational structures, for example, syn-C6H10O has 112 structures and the transition state of E-6c-TS has 182 distinguishable structures. The numbers of structures for other species (reactants and transition states) are given in the Supplementary Information. Multiple structures are a form of anharmonicity since a harmonic potential leads to only a single structure. Our kinetics calculations include all distinguishable structures and the anharmonicity that they generate; we use the MS-T method23 to treat the multiple-structure anharmonicity, torsional-potential anharmonicity, and vibration–rotation coupling. The effect of these anharmonicities on the reaction rates is shown in Supplementary Table 6; over the 190–298 K temperature range, it varies from a factor of 0.025 to a factor of 0.74 for the bicyclic ring closure reactions, which corresponds to decreasing the rate by as much as a factor of 40 as compared to the quasiharmonic approximation. The effects on the rate are smaller—but not negligible—for the other kinds of reaction, varying between factors of 0.55 and 1.57 for the hydrogen shift reactions and between factors of 0.27 and 1.28 for dioxirane cyclization reactions. In the rest of the discussion, with one exception (Z-5hs-TS2, which is explained below), we discuss only the lowest-energy structure of each species.
The unimolecular reactions of syn-C5H8O3
The bicyclic ring closure reaction we find for syn-C5H8O3 is labeled above as Z-5rc and is shown Fig. 3, which also shows the enthalpy profile at 0 K (i.e., the summation of electronic energy and the zero-point vibrational energy) computed by CCSD(T)-F12a/jun-cc-pVTZ based on MN15-L/MG3S geometries. The reaction is analogous to the bimolecular reaction of CH2OO with HCHO24. The previous assumption would have been that the dominant unimolecular reaction of this syn-Criegee intermediates would be the 1,4-hydrogen shift process that we have labeled Z-5hs, which is also shown in Fig. 3. The transition states for the hydrogen shift reactions have both an equatorial structure and an axial structure: Z-5hs-TS1 (shown in Fig. 3) and Z-5hs-TS2 (shown in Supplementary Fig. 1). The equatorial structure has a lower energy.

The calculated enthalpy profile of syn-C5H8O3
The bicyclic ring closure reaction involves the concerted formation of two bonds; it involves binding of the carbonyl carbon atom of the CHO group to the terminal oxygen atom of the HCOO group and binding of the carbonyl oxygen atom of the HCO group to the carbon atom of the HCOO group. This process is similar to the unimolecular reactions of other Criegee intermediates in the ozonolysis of isoprene and β-myrcene25,26,27, where the HCOO group attacks the C=C double bond. However, the enthalpies of activation at 0 K are above 10 kcal/mol for the similar unimolecular reactions of the HCOO group with the C=C group;25,26,27,28,29 this is also qualitatively consistent with the corresponding CH2OO+HCHO and CH2OO+C2H4 reactions, where the enthalpy of activation at 0 K for the CH2OO+C2H4 reaction is higher than that of the CH2OO+HCHO reaction24,30. There are two different transition state structures for SOZ formation in syn-C5H8O3 (see Supplementary Fig. 2). Here, we only consider the lowest transition state Z-5rc-TS because Z-5rc-TS is 3.37 kcal/mol lower than Z-5rc-TS1 (see Supplementary Fig. 2). The two C–O bond distances of the new bonds at the transition state (Z-5rc-TS) are respectively 2.11 and 2.15 Å, which may be compared to values of 1.45 and 1.42 Å in the product.
Table 1 shows that the enthalpy of activation (all enthalpies of reaction and enthalpies of activation in the text are given for 0 K to illustrate the energetics in the well-defined low-temperature limit, but the results are similar at atmospheric temperatures; activation energies at atmospheric temperatures are given in the SI) for the SOZ-forming bicyclic ring closure as calculated by CCSD(T)-F12a/jun-cc-pVTZ//MN15-L/MG3S is 2.27 kcal/mol (see Table 1) and that this is lower than that of the hydrogen shift process by about 14.5 kcal/mol (i.e., 16.73−2.27). Moreover, Fig. 3 shows that the product Z-5rc-P of the bicyclic ring closure reaction is much more stable than the product Z-5hs-P1 of the hydrogen shift reaction because the enthalpy of reaction for Z-5rc is 18.4 kcal/mol lower (i.e., 33.17−14.81). The present finding is different from the previously reported results which indicated a low barrier for SOZ formation only for the case of six carbons in the product ring31,44.
To further check the reliability of the CCSD(T) enthalpy of activation, we performed two higher-order calculations, and the results of these are shown in Table 1. The quite different very-high-level W2X32 and WMS33 methods almost give identical results for Z-5rc; this shows it is not required to carry out post-CCSD(T) calculations on the bicyclic ring closure reaction of the large-sized Criegee intermediate syn-C5H8O3, and the good agreement of activation enthalpies in Table 1 shows that we can trust the CCSD(T) calculation for further studies. A further indication that post-CCSD(T) theory is not needed is that the [E(CCSDT(Q))−E(CCSD(T))] contribution to the Z-5rc barrier height calculated with the VDZ(d)32 basis is only 0.07 kcal/mol (Supplementary Table 1).
The WMS, W2X, and CCSD(T) calculations in Table 1 are too expensive for direct dynamics calculations. We selected the MN15-L density functional for the present work because of its good performance demonstrated previously for strongly correlated systems and barrier heights34 and based on its successful use in our previous investigations of small Criegee intermediates35,36. The results in Table 1 then provide further confirmation of its usefulness for the present problem. In particular, the MN15-L/MG3S and MN15-L/maug-cc-pVTZ density functional calculations provide enthalpies of activation of respectively 2.8 and 2.3 kcal/mol for Z-5rc, in good agreement with the high-level WMS value of 2.5 kcal/mol; this shows more specifically that MN15-L/MG3S can provide useful results for describing the bicyclic ring closure reactions of Criegee intermediates. This is important because MN15-L is less expensive and can be used for full rate constant calculations. Thus, MN15-L/MG3S is selected for the direct dynamics components of our rate constant calculations.
The unimolecular reactions of anti-C5H8O3
The enthalpy of activation for isomerization of anti-C5H8O3 to syn-C5H8O3 is 35.43 kcal/mol as shown in Supplementary Fig. 3. The isomerization mechanism between anti-C5H8O3 and syn-C5H8O3 is expected to be similar to that in small-sized Criegee intermediates28,35,37. Thus, syn-C5H8O3 and anti-C5H8O3 act as independent reactants.
A bicyclic ring closure reaction is also found for anti-C5H8O3, as seen in Fig. 4 (which also shows the enthalpy profile at 0 K computed by CCSD(T)-F12a/jun-cc-pVTZ//MN15-L/MG3S); however, it has a much higher barrier in this case. Furthermore, there is only one transition state structure for SOZ formation in anti-C5H8O3. Table 1 shows that that the enthalpies of activation for E-5rc and E-5c are both calculated to be 13.7 kcal/mol; this indicates that the E-5rc reaction, although not dominant, should not be neglected in computing the removal rate of anti-C5H8O3.

The calculated enthalpy profile of anti-C5H8O3 by CCSD(T)-F12a/jun-cc-pVTZ
The unimolecular reactions of syn-C6H10O3 and anti-C6H10O3
We also investigate the syn- and anti-Criegee intermediates from the ozonolysis of cyclic C6H10. Supplementary Figure 4 shows that the dominant reaction pathway of syn-C6H10O3 is similar to the unimolecular reaction of syn-C5H8O3. The activation enthalpy for Z-6rc is calculated to be 1.5 kcal/mol by CCSD(T)-F12a/jun-cc-pVTZ//MN15-L/MG3S, which is about 14.4 kcal/mol lower than that of the previously suggested38 hydrogen transfer shift Z-6hs-TS1 in Table 1; this means that this pathway is much faster than that of the hydrogen shift. However, the previous theoretical results reported a much higher barrier of 5.1 (ref. 6) or 8.5 kcal/mol39 for the bicyclic ring closure reactions of syn-C6H8O3. A key issue in this discrepancy is that one must find the lowest-energy transition state, and there are four different transition state structures for SOZ formation from syn-C6H10O3 (see Supplementary Fig. 5) and two different transition state structures for the bicyclic ring closure reaction in syn-C5H8O3; this situation occurs because each of the two central CH2 groups in syn-C6H10O3 has two different pathways leading to SOZ formation; in comparison, the one central CH2 group in syn-C5H8O3 leads to two pathways. This example illustrates how there are more transition state structures for SOZ formation and how one locates more transition state structures when the carbon chain becomes longer. The lowest-energy transition state Z-6rc-TS is considered here because the CCSD(T)-F12a/jun-cc-pVTZ//MN15-L/MG3S calculations indicate that it is 1.97 kcal/mol lower than the second lowest structure, 3.89 kcal/mol lower than the third lowest structure, and 7.68 kcal/mol lower than the fourth lowest structure (see Supplementary Fig. 5).
The enthalpy of activation for isomerization of syn-C6H10O3 to anti-C6H10O3 is 37.37 kcal/mol (see Supplementary Fig. 6); this shows that syn-C6H10O3 and anti-C6H10O3 can be considered to be independent reactants.
For anti-C6H10O3, there are two different transition state structures for SOZ formation (see Supplementary Fig. 7). We consider E-6rc-TS because E-6rc-TS is 3.41 kcal/mol lower than E-6rc-TS1 (see Supplementary Fig. 7). Table 1 and Supplementary Fig. 8 show an enthalpy of activation of 4.8 kcal/mol for bicyclic ring closure E-6rc, which is 9.5 kcal/mol lower than that of the dioxirane cyclization process E-6c-TS. The enthalpy of activation at 0 K for E-6rc is about 9 kcal/mol lower than that of E-5rc because of different chain strain between C5 and C6 Criegee intermediates. Thus, for the C6 case, this reaction dominates the previously known reactions for both syn and anti.
Cyclic C6H10 represents the skeleton of monoterpenes such as limonene, 2-carene, 3-carene, phellandrene, terpinene, and terpinolene17. Therefore, the present conclusions are expected to be important for the monoterpenes; however, the SOZ-forming bicyclic ring closure was previously only included in the atmospheric modeling of the ozonolysis of β-caryophyllene40.
Kinetics
The calculated rate constants are given in Table 2 and Supplementary Table 3. The unimolecular rate constants were fitted using the following formula:
The fitted parameters are provided in Supplementary Table 4. Table 2 shows that the SOZ-forming bicyclic ring closures Z-5rc and Z-6rc of syn-C5H8O3 and C6H10O3 are about 105–109 times faster than the previously suggested hydrogen transfer process (Z-5hs and Z-6hs) and the SOZ-forming bicyclic ring closure E-6rc is about 106–108 times faster than the previously suggested dioxirane cyclization reaction.
Supplementary Table 5 gives the temperature-dependent activation energies Ea (computed as local slopes of Arrhenius plots41). Over the atmospherically relevant temperature range 190–298 K, the activation energies for the ring closure reactions increase as the temperature is lowered; for example, Ea for Z-5rc increases from 1.1 to 2.0 kcal/mol.
In Table 2, the rate constants of Z-5rc are much faster than those of Z-6rc, while the enthalpies of activation for Z-5rc at 0 K is 0.42 kcal/mol higher than that of Z-6rc calculated by WMS (Table 1); this is mainly caused by the torsional anharmonicity (see Supplementary Table 6). For example, torsional anharmonicity decreases the rate constant at 298 K by a factor of 2 for Z-5rc and by a factor of 40 for Z-6rc. Therefore, the torsional anharmonicity can play important roles in determining the kinetics of bicyclic ring closure reactions of Criegee intermediates, and this conclusion extends to large systems as well as the present systems.
Implications for atmospheric chemistry
Although SOZ formation is well known in the literature6,31,38,39,40,42,43,44 and previous investigation indicated that SOZ formation dominates over the hydrogen shift for the syn SCI from cycloalkenes ozonlysis6, the rates of reaction of large SCIs are unclear in previous work because of the different barrier heights for SOZ formation reported in the literature for the same Criegee intermediate. In particular, the barrier height was reported to be 5.1 (ref. 6) or 8.5 (ref. 39) kcal/mol for secondary ozonide formation in the syn-Criegee intermediate from cyclohexene ozonolysis. The primary objective of the present article was to provide data on the reaction rates for producing SOZs in gas-phase reactions of SCIs. The present calculations are for collisonally stabilized reactants, and we note that pressure-dependent kinetics calculations not reported here would be required to to fully understand and predict the extent of collisional stabilization of these large Criegee intermediates; future work to study this could be carried out using master equation simulations45 or the system-specific quantum Rice–Ramsperger–Kassel model46.
Unimolecular reactions of small Criegee intermediates, such as CH2OO35,47,48,49 syn and anti-CH3CHOO35,37,50,51 (CH3)2COO36,52 and syn/anti-C2H5CHOO28,29, were previously investigated by using both theoretical and experimental methods. The generally accepted view from these studies is that syn-Criegee intermediates react mainly by hydrogen shift (hs) reactions. Hydrogen shift reactions of Criegee intermediates from a methyl or ethyl group to the terminal oxygen atom are especially important because they lead to the formation of OH radical11,36,50,51,53 and can be the initial step for the formation of highly oxygenated molecules, which make a significant contribution to the formation of atmospheric organic aerosols54,55. The dominant pathway of small anti-Criegee intermediates is usually assumed to be cyclization (c) to a dioxirane.35,37 The enthalpies of activation of the unimolecular reactions of small Criegee intermediates are in the range between 15 and 30 kcal/mol35,36,37; this implies that the unimolecular reactions of small Criegee intermediates such as CH2OO and anti-CH3CHOO cannot compete well with bimolecular reaction with water, water dimer, SO2, or atmospheric acids10,35,56.
The bicyclic ring closure reactions that we report here are important for understanding the atmospheric lifetimes of large Criegee intermediates—not only for those from the ozonolysis of C5H8 and C6H10 but also for a wider class of compounds. While previous experimental and theoretical results have suggested that secondary ozonides (SOZ) are formed by unimolecular reactions in the ozonolysis of cycloalkenes6,40, the dominant pathways are generally assumed to be the same as those for the small Criegee intermediates. For example, recent studies of the ozonolysis of limonene, α-pinene, other monoterpenes, and indene included the hydrogen shift reaction as the only unimolecular step in the mechanisms57,58,59. Since the structural features of large Criegee intermediates can be quite different from those of small Criegee intermediates, it is important to understand whether they react by different mechanisms.
Previous investigations have shown that the unimolecular reactions of the simple Criegee intermediates occurring via hydrogen shifts lead to the formation of OH radical via the decomposition of -OOH groups11,50,51,53. However, our calculated results show that the unimolecular reactions of larger syn-Criegee intermediates from the ozonolysis of C5H8 and C6H10 predominantly undergo reactions in which no −OOH group is formed. Thus, the reactions limit OH production in the ozonolysis of C5H8 and C6H10; this further leads to the reduction of the oxidation capacity of the corresponding Criegee intermediates from cyclic C6 structures.
The reaction in three of the Criegee intermediates (syn-C5H8O3, syn/anti-C6H10O3) is so fast that the atmospheric lifetimes of three Criegee intermediates are much less than 10−5 s. However, the atmospheric lifetimes associated with the corresponding bimolecular reactions such as reactions with SO2, H2O, water dimer, and other atmospheric acids are often more than 10−3 s56. We conclude that large Criegee intermediates that are produced by ozonolysis do not act as potential oxidants to oxidize SO2 to SO3. This is especially important for monoterpenes with structural features similar to C6H10.
The formation of highly oxidized molecules and radicals in the ozonolysis of cyclic monoterpenes plays a significant role in atmospheric organic aerosol formation55. Experimental results have shown that highly oxidized molecules and radicals can be formed in the ozonolysis of cyclohexene and limonene38,57. However, our results presented here clearly show that the dominant sink of syn- and anti-Criegee intermediates from six-carbon-atom cyclic monoterpenes is not the hydrogen shift mechanism as suggested in the literature; 38,57 Thus, the present investigation upsets one aspect of conventional thinking about the formation of highly oxidized molecules and radicals.
The reactions studied here are important in illustrating a more general phenomenon, namely that large Criegee intermediates may have lower energy unimolecular pathways that are analogous to the known bimolecular reactions of Criegee intermediates. For example, the ozonolysis of fatty acids20 can lead to Criegee intermediates with an COOH group, and these could react by a mechanism similar to the bimolecular reaction of Criegee intermediates with atmospheric acids with barrierless reactions60,61. Thus, the present results stimulate one to consider faster unimolecular reaction mechanisms of large Criegee intermediates.
Summary
In the unimolecular reactions of Criegee intermediates, the accepted reaction mechanisms are hydrogen shifts and dioxirane cyclization. Here, we used electronic structure calculations and multistructural variational transition state theory with small-curvature tunneling to explore unimolecular reactions of large-sized stabilized Criegee intermediates produced in the ozonolysis of cyclic monoterpenes. We find that multistructural effects are important for determining the barrier heights and torsion anharmonicity.
For large syn-Criegee intermediates from the ozonolysis of cyclic monoterpenes, we find a mechanism involving unimolecular reactions with enthalpies of activation about 2–3 kcal/mol. We find that the unimolecular rate constants of the processes are 106–109 s−1 in the temperature range between 190 and 298 K. We find for syn-Criegee intermediates that a low-energy path for internal SOZ formation is available even with only five carbons in the product ring, whereas previous work44 incorrectly indicated that a minimum of six carbons is required.
We conclude that unexpectedly fast unimolecular reactions of Criegee intermediates significantly limit the oxidation capacity of Criegee intermediates, and we suggest that the reaction types with low-energy barriers as reported here should be widespread.
Methods
Computation of rate constants
Rate constants (k) were calculated by a dual-level approximation:
where level 1 (L1) of electronic structure employs the MN15-L local meta exchange-correlation functional34 with the MG3S basis set62, level 2 (L2) employs WMS, \(V^\ddagger\) is the classical barrier height, R is the gas constant, T is the temperature, and MS-CVT/SCT is multistructural63 canonical variational theory64,65 with small-curvature tunneling66.
The conformational structures of the reactants and transition states were calculated by multistructural method with coupled torsional-potential anharmonicity (MS-T)23. The frequencies were scaled by the factor of 0.973 for MN15-L/MG3S as obtained in a standard way67 to account for vibrational anharmonicity and systematic errors in the electronic structure.
Electronic structure methods
Our previous investigations have shown that the MN15-L functional can provide useful accuracy for the unimolecular reactions of small and medium-sized Criegee intermediates35,36. Our previous work also showed that obtaining quantitative rate constants for the unimolecular reactions of Criegee intermediates requires calculations beyond CCSD(T)35,36, but here we employ very-high-level calculations to show that CCSD(T) is sufficient for the low-energy pathways identified in this work. In addition, the composite methods WMS and W2X were used to do single-point calculations to show the reliability of the method chosen for direct dynamics. Even without the dynamics calculations, these high-level calculations of barrier heights would already show the importance of the bicyclic ring closure reactions.
It is impossible to put quantitative error bars on the electronic structure and rate constant calculations, but some discussion of reliability may be useful. Based on previous work, the uncertainties in the rate constant calculations are expected to be dominated by the electronic structure calculations rather than the errors in the dynamical parts of the calculations68. In electronic structure calculations, there are three factors to be considered: optimized geometries, vibrational frequencies, and single-point electronic energies69. In a previous paper, we have shown good agreement of CCSD(T) and MN15-L for optimized geometries and zero-point vibrational energies on similar systems35. Based on these tests and the previous work showing MN15-L is able to yield reasonably accurate vibrational frequencies34, the error of the electronic structure calculations is most likely not dominated by geometry errors or frequency errors. Previous experience with the methods used here indicates that the single-point electronic energies have typical errors of 1 kcal/mol or less32,34,70; this would cause errors in 300 K rate constants of a factor of 5 or less. This is consistent with previous work where we found that MN15-L yielded rate constants only a factor of 2 different from experimental results for the hydrogen shift of syn-CH3CHOO35, and it provided results consistent with experimental data in the hydrogen shift of (CH3)2COO36
Data Availability
All data analyzed during this study are included in this published article and its supplementary information files. Supplementary information is available in the online version of the paper. Reprints and permissions information are available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to B.L., J.L.B., and D.G.T.
Code Availability
The calculations were carried out using the Polyrate, Gaussrate, and MSTor computer codes that are available at no cost at http://truhlar.chem.umn.edu/content/software
References
Criegee, R. Mechanism of ozonolysis. Angew. Chem. Int. Ed. Engl. 14, 745–752 (1975).
Johnson, D. & Marston, G. The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere. Chem. Soc. Rev. 37, 699–716 (2008).
Mauldin III, R. L. et al. A new atmospherically relevant oxidant of sulphur dioxide. Nature 488, 193–196 (2012).
Osborn, D. L. & Taatjes, C. A. The physical chemistry of Criegee intermediates in the gas phase. Int. Rev. Phys. Chem. 34, 309–360 (2015).
Zhong, J., Kumar, M., Francisco, J. S. & Zeng, X. C. Insight into chemistry on cloud/aerosol water surfaces. Acc. Chem. Res. 51, 1229–1237 (2018).
Chuong, B., Zhang, J. & Donahue, N. M. Cycloalkene ozonolysis: collisionally mediated mechanistic branching. J. Am. Chem. Soc. 126, 12363–12373 (2004).
Vereecken, L., Glowacki, D. R. & Pilling, M. J. Theoretical chemical kinetics in tropospheric chemistry: methodologies and applications. Chem. Rev. 115, 4063–4114 (2015).
Lee, Y.-P. Perspective: Spectroscopy and kinetics of small gaseous Criegee intermediates. J. Chem. Phys. 143, 020901 (2015).
Su, Y.-T. et al. Extremely rapid self-reaction of the simplest Criegee intermediate CH2OO and its implications in atmospheric chemistry. Nat. Chem. 6, 477–483 (2014).
Chao, W., Hsieh, J.-T., Chang, C.-H. & Lin, J. J.-M. Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor. Science 347, 751–754 (2015).
Liu, F., Beames, J. M., Petit, A. S., McCoy, A. B. & Lester, M. I. Infrared-driven unimolecular reaction of CH3CHOO Criegee intermediates to OH radical products. Science 345, 1596–1598 (2014).
Taatjes, C. A. et al. Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO. Science 340, 177–180 (2013).
Su, Y.-T., Huang, Y.-H., Witek, H. A. & Lee, Y.-P. Infrared absorption spectrum of the simplest Criegee intermediate CH2OO. Science 340, 174–176 (2013).
Taatjes, C. A. et al. Direct observation of the gas-phase Criegee intermediate (CH2OO). J. Am. Chem. Soc. 130, 11883–11885 (2008).
Beames, J. M., Liu, F., Lu, L. & Lester, M. I. Ultraviolet spectrum and photochemistry of the simplest Criegee intermediate CH2OO. J. Am. Chem. Soc. 134, 20045–20048 (2012).
Jr-Min Lin, J. & Chao, W. Structure-dependent reactivity of Criegee intermediates studied with spectroscopic methods. Chem. Soc. Rev. 46, 7483–7497 (2017).
Atkinson, R. & Arey, J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review. Atmos. Environ. 37, 197–219 (2003).
Mellouki, A., Le Bras, G. & Sidebottom, H. Kinetics and mechanisms of the oxidation of oxygenated organic compounds in the gas phase. Chem. Rev. 103, 5077–5096 (2003).
Rossignol, S. et al. Atmospheric photochemistry at a fatty acid–coated air-water interface. Science 353, 699–702 (2016).
Kawamura, K. & Gagosian, R. B. Implications of ω-oxocarboxylic acids in the remote marine atmosphere for photo-oxidation of unsaturated fatty acids. Nature 325, 330 (1987).
Vereecken, L. & Peeters, J. The 1,5-H-shift in 1-butoxy: a case study in the rigorous implementation of transition state theory for a multirotamer system. J. Chem. Phys. 119, 5159–5170 (2003).
Yu, T., Zheng, J. & Truhlar, D. G. Multi-structural variational transition state theory. Kinetics of the 1,4-hydrogen shift isomerization of the pentyl radical with torsional anharmonicity. Chem. Sci. 2, 2199–2213 (2011).
Zheng, J. & Truhlar, D. G. Quantum thermochemistry: multistructural method with torsional anharmonicity based on a coupled torsional potential. J. Chem. Theory Comput. 9, 1356–1367 (2013).
Aplincourt, P. & Ruiz-López, M. F. Theoretical investigation of reaction mechanisms for carboxylic acid formation in the atmosphere. J. Am. Chem. Soc. 122, 8990–8997 (2000).
Kuwata, K. T., Valin, L. C. & Converse, A. D. Quantum chemical and master equation studies of the methyl vinyl carbonyl oxides formed in isoprene ozonolysis. J. Phys. Chem. A 109, 10710–10725 (2005).
Kuwata, K. T. & Valin, L. C. Quantum chemical and RRKM/master equation studies of isoprene ozonolysis: methacrolein and methacrolein oxide. Chem. Phys. Lett. 451, 186–191 (2008).
Deng, P., Wang, L. & Wang, L. Mechanism of gas-phase ozonolysis of β-myrcene in the atmosphere. J. Phys. Chem. A 122, 3013–3020 (2018).
Yin, C. & Takahashi, K. How does substitution affect the unimolecular reaction rates of Criegee intermediates? Phys. Chem. Chem. Phys. 19, 12075–12084 (2017).
Vereecken, L., Novelli, A. & Taraborrelli, D. Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates. Phys. Chem. Chem. Phys. 19, 31599–31612 (2017).
Vereecken, L., Harder, H. & Novelli, A. The reactions of Criegee intermediates with alkenes, ozone, and carbonyl oxides. Phys. Chem. Chem. Phys. 16, 4039–4049 (2014).
Kuwata, K. T., Kujala, B. J., Morrow, Z. W. & Tonc, E. Quantum chemical and RRKM/master equation studies of cyclopropene ozonolysis. Comput. Theor. Chem. 965, 305–312 (2011).
Chan, B. & Radom, L. W2X and W3X-L: cost-effective approximations to W2 and W4 with kJ mol–1 accuracy. J. Chem. Theory Comput. 11, 2109–2119 (2015).
Zhao, Y. et al. Extrapolation of high-order correlation energies: the WMS model. Phys. Chem. Chem. Phys. 20, 27375–27384 (2018).
Yu, H. S., He, X. & Truhlar, D. G. MN15-L: a new local exchange-correlation functional for Kohn–Sham density functional theory with broad accuracy for atoms. Mol. solids J. Chem. Theory Comput. 12, 1280–1293 (2016).
Long, B., Bao, J. L. & Truhlar, D. G. Atmospheric chemistry of Criegee intermediates: unimolecular reactions and reactions with water. J. Am. Chem. Soc. 138, 14409–14422 (2016).
Long, B., Bao, J. L. & Truhlar, D. G. Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere. Proc. Natl. Acad. Sci. USA 115, 6135–6140 (2018).
Kuwata, K. T., Hermes, M. R., Carlson, M. J. & Zogg, C. K. Computational studies of the isomerization and hydration reactions of acetaldehyde oxide and methyl vinyl carbonyl oxide. J. Phys. Chem. A 114, 9192–9204 (2010).
Newland, M. J. et al. The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling. Atmos. Chem. Phys. 18, 6095–6120 (2018).
Monge-Palacios, M., Rissanen, M. P., Wang, Z. & Sarathy, S. M. Theoretical kinetic study of the formic acid catalyzed Criegee intermediate isomerization: multistructural anharmonicity and atmospheric implications. Phys. Chem. Chem. Phys. 20, 10806–10814 (2018).
Khan, M. A. H. et al. A modeling study of secondary organic aerosol formation from sesquiterpenes using the STOCHEM global chemistry and transport model. J. Geophys. Res. Atmos. 122, 4426–4439 (2017).
Truhlar, D. G. & Gray, J. C. Interpretation and emperature dependence of the energy of activation for the reactions H+Cl2, H2 + I, H + H2, and isotopic analogs. Chem. Phys. Lett. 57, 93–99 (1978).
Hatakeyama, S., Kobayashi, H. & Akimoto, H. Gas-phase oxidation of sulfur dioxide in the ozone-olefin reactions. J. Phys. Chem. 88, 4736–4739 (1984).
Nguyen, T. L. et al. The gas-phase ozonolysis of β-caryophyllene (C15H24). Part II: a theoretical study. Phys. Chem. Chem. Phys. 11, 4173–4183 (2009).
Vereecken, L. & Francisco, J. S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 41, 6259–6293 (2012).
Glowacki, D. R., Liang, C.-H., Morley, C., Pilling, M. J. & Robertson, S. H. MESMER: an open-source master equation solver for multi-energy well reactions. J. Phys. Chem. A 116, 9545–9560 (2012).
Bao, J. L., Zheng, J. & Truhlar, D. G. Kinetics of hydrogen radical reactions with toluene including chemical activation theory employing system-specific quantum RRK theory calibrated by variational transition state theory. J. Am. Chem. Soc. 138, 2690–2704 (2016).
Berndt, T. et al. Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone under atmospheric conditions. Phys. Chem. Chem. Phys. 17, 19862–19873 (2015).
Chhantyal-Pun, R., Davey, A., Shallcross, D. E., Percival, C. J. & Orr-Ewing, A. J. A kinetic study of the CH2OO Criegee intermediate self-reaction, reaction with SO2 and unimolecular reaction using cavity ring-down spectroscopy. Phys. Chem. Chem. Phys. 17, 3617–3626 (2015).
Newland, M. J. et al. Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions. Phys. Chem. Chem. Phys. 17, 4076–4088 (2015).
Kidwell, N. M., Li, H., Wang, X., Bowman, J. M. & Lester, M. I. Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products. Nat. Chem. 8, 509–514 (2016).
Green, A. M., Barber, V. P., Fang, Y., Klippenstein, S. J. & Lester, M. I. Selective deuteration illuminates the importance of tunneling in the unimolecular decay of Criegee intermediates to hydroxyl radical products. Proc. Natl. Acad. Sci. USA 114, 12372–12377 (2017).
Drozd, G. T., Kurtén, T., Donahue, N. M. & Lester, M. I. Unimolecular decay of the dimethyl-substituted Criegee intermediate in alkene ozonolysis: decay time scales and the importance of tunneling. J. Phys. Chem. A 121, 6036–6045 (2017).
Lester, M. I. & Klippenstein, S. J. Unimolecular decay of Criegee intermediates to OH radical products: prompt and thermal decay processes. Acc. Chem. Res. 51, 978–985 (2018).
Bianchi, F. et al. New particle formation in the free troposphere: a question of chemistry and timing. Science 352, 1109–1112 (2016).
Tröstl, J. et al. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature 533, 527–531 (2016).
Khan, M. A. H., Percival, C. J., Caravan, R. L., Taatjes, C. A. & Shallcross, D. E. Criegee intermediates and their impacts on the troposphere. Environ. Sci. Process. Impacts 20, 437–453 (2018).
Rissanen, M. P. et al. The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene. J. Am. Chem. Soc. 136, 15596–15606 (2014).
Jokinen, T. et al. Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere. Angew. Chem. Int. Ed. 53, 14596–14600 (2014).
Chiappini, L. et al. Secondary organic aerosol formation from aromatic alkene ozonolysis: influence of the precursor structure on yield, chemical composition, and mechanism. J. Phys. Chem. A 123, 1469–1484 (2019).
Long, B., Cheng, J.-R., Tan, X.-f & Zhang, W.-j Theoretical study on the detailed reaction mechanisms of carbonyl oxide with formic acid. J. Mol. Struct. THEOCHEM 916, 159–167 (2009).
Welz, O. et al. Rate coefficients of C1 and C2 Criegee intermediate reactions with formic and acetic acid near the collision limit: direct kinetics measurements and atmospheric implications. Angew. Chem. Int. Ed. 53, 4547–4550 (2014).
Lynch, B. J., Zhao, Y. & Truhlar, D. G. Effectiveness of diffuse basis functions for calculating relative energies by density functional theory. J. Phys. Chem. A 107, 1384–1388 (2003).
Bao, J. L., Meana-Pañeda, R. & Truhlar, D. G. Multi-path variational transition state theory for chiral molecules: the site-dependent kinetics for abstraction of hydrogen from 2-butanol by hydroperoxyl radical, analysis of hydrogen bonding in the transition state, and dramatic temperature dependence of the activation energy. Chem. Sci. 6, 5866–5881 (2015).
Garrett, B. C. & Truhlar, D. G. Criterion of minimum state density in the transition state theory of bimolecular reactions. J. Chem. Phys. 70, 1593–1598 (1979).
Bao, J. L. & Truhlar, D. G. Variational transition state theory: theoretical framework and recent developments. Chem. Soc. Rev. 46, 7548–7596 (2017).
Liu, Y.-P. et al. Molecular modeling of the kinetic isotope effect for the [1,5]-sigmatropic rearrangement of cis-1,3-pentadiene. J. Am. Chem. Soc. 115, 2408–2415 (1993).
Alecu, I. M., Zheng, J., Zhao, Y. & Truhlar, D. G. Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theory Comput. 6, 2872–2887 (2010).
Klippenstein, S. J., Pande, V. & Truhlar, D. G. Chemical kinetics and mechanisms of complex systems: a perspective on recent theoretical advances. J. Am. Chem. Soc. 136, 528–546 (2014).
Long, B., Bao, J. L. & Truhlar, D. G. Kinetics of the strongly correlated CH3O + O2 reaction: the importance of quadruple excitations in atmospheric and combustion chemistry. J. Am. Chem. Soc. 141, 611–617 (2019).
Zheng, J., Zhao, Y. & Truhlar, D. G. The DBH24/08 database and its use to assess electronic structure model chemistries for chemical reaction barrier heights. J. Chem. Theory Comput. 5, 808–821 (2009).
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (41775125), by the Science and Technology Foundation of Guizhou Province, China ([2018]1080), and by the Science and Technology Foundation of Guizhou Provincial Department of Education, China ([2015]350), and the U.S. Department of Energy (DE-SC0015997). Computations were performed using resources of Minnesota Supercomputing Institute and the National Energy Research Scientific Computing Center.
Author information
Authors and Affiliations
Contributions
B.L. and J.L.B. performed the calculations; B.L., J.L.B., and D.G.T. conceived and designed the calculations, analyzed the data, and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal Peer Review Information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Long, B., Bao, J.L. & Truhlar, D.G. Rapid unimolecular reaction of stabilized Criegee intermediates and implications for atmospheric chemistry. Nat Commun 10, 2003 (2019). https://doi.org/10.1038/s41467-019-09948-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-09948-7
This article is cited by
-
The atmospheric relevance of primary alcohols and imidogen reactions
Scientific Reports (2023)
-
Open questions on the reactivity of Criegee intermediates
Communications Chemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
