
Abstract
Understanding the growth pathway of faceted alloy nanoparticles at the atomic level is crucial to morphology control and property tuning. Yet, it remains a challenge due to complexity of the growth process and technical limits of modern characterization tools. We report a combinational use of multiple cutting-edge in situ techniques to study the growth process of octahedral Pt3Ni nanoparticles, which reveal the particle growth and facet formation mechanisms. Our studies confirm the formation of octahedral Pt3Ni initiates from Pt nuclei generation, which is followed by continuous Pt reduction that simultaneously catalyzes Ni reduction, resulting in mixed alloy formation with moderate elemental segregation. Carbon monoxide molecules serve as a facet formation modulator and induce Ni segregation to the surface, which inhibits the (111) facet growth and causes the particle shape to evolve from a spherical cluster to an octahedron as the (001) facet continues to grow.
Similar content being viewed by others

Introduction
Faceted platinum alloy nanoparticles, in particular octahedral Pt–Ni, have been recognized as one of the most promising cathode catalysts for use in hydrogen fuel cells for their dramatically improved oxygen reduction reaction (ORR) activity compared to their spherical counterparts and pure Pt1,2,3,4. These findings revealed significant catalytic structure-property correlations and suggested the importance of controlling the facet growth of nanoparticles to achieve the desired property. Although considerable success has been achieved in synthesizing faceted alloy nanoparticles after years of intensive research, these achievements were mostly empirical. Knowledge of the growth pathway, which is essential to guide rational synthesis, is still lacking. The past decade has witnessed technological advances of in situ electron microscopy and its use in obtaining the growth trajectory of pure metal nanoparticles5,6,7,8,9,10,11,12. Compared with pure metals, the growth pathway of faceted alloy nanoparticles is more complicated. With multiple elements being involved, it is challenging to identify their behaviors, both individually as different elements and collectively as alloy components, during the particle growth13,14.
Herein we report a combinational use of several cutting-edge in situ characterization techniques, including aberration-corrected scanning transmission electron microscopy (AC-STEM), ambient pressure X-ray photoelectron spectroscopy (AP-XPS), and X-ray absorption spectroscopy (XAS), to study the growth process of octahedral Pt3Ni nanoparticles. By integrating the information obtained from these techniques, together with computational simulations, a clear pathway for the particle growth and facet formation is revealed.
Results
In situ STEM characterizations
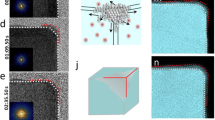
Direct visualization of the growth process of octahedral Pt3Ni was performed on a JEOL-JEM 3100R05 microscope that is equipped with a Protochips AtmosphereTM gas cell system (Supplementary Fig. 1)15,16, realizing dynamic observation at the atomic scale under atmospheric pressures. The reaction condition for growing octahedral Pt3Ni followed a solid-state chemistry method we recently developed, in which H2 acts as a reductant and CO acts as a capping agent for morphology control1. Figure 1a shows the time-sequenced STEM images of one single particle at its different growth stages (top row). Simulated images, presented in the bottom row, were obtained based on the experimental data. It was quite challenging to image the particles clearly at their early growth stage (denoted as t0 = 0 s), primarily because of their high structural instability and mobility in the small cluster size range. Moreover, the use of low-dose STEM imaging condition in this study, which was necessary for minimizing the electron beam effect, sacrificed the image quality to some extent (Supplementary Note 1, Supplementary Fig. 2). Nevertheless, the observed sphere-like characteristic indicated no apparent facet formation of the particles and highly dynamic property in their early growth stage, which has been discussed in supplementary materials (Supplementary Note 2). Facet formation and distinct lattice fringes were observed to evolve when the particle size slightly exceeded 1 nm. Two groups of facets, i.e., {111} and {001}, were indexed as indicated by the atomic model in Fig. 1b. Noticeably, the {111} facets exhibited a significantly slower growth rate compared with the {001} under the reaction condition, which was evidenced by a smaller number of atomic layers along the [111] axis than along the [001] (Fig. 1c). Our previous study has identified the important role of CO molecules in inhibiting the Pt3Ni {111} growth1, which led to the formation of octahedral morphology rather than a spheroid one. The slower {111} growth resulted in the gradual domination of this group of facet planes and diminishment of the {001} over the reaction time (Fig. 1d, Supplementary Fig. 3), which was accompanied by a continuous morphological change of the growing particle. The in situ observation of other growing particles found a same trend (Supplementary Figs. 4–6) with a homogenous distribution of the particles during the growth (Supplementary Fig. 7) and a high yield of octahedral morphology in the final product (Supplementary Fig. 8), suggesting this is a generic pathway for octahedral Pt3Ni growth (a more detailed discussion can be found in the Supplementary Note 2, Supplementary Fig. 9). Thus we constructed three-dimensional (3D) models based on the in situ STEM data to better illustrate the growth process of octahedral Pt3Ni (Fig. 1e), when there is a continual increase in size along with a gradual morphological transition from spherical cluster to polyhedron and finally to octahedron.

Shape evolution of the growing Pt3Ni nanoparticle. a In situ STEM images of one growing Pt3Ni nanoparticle (upper row) and corresponding 2D projection model (lower row), scale bar: 1 nm. b Representative atomic model (not to scale) of the nanoparticle during growth. c Number of grown layers of the (001) and (111) facets (Error bars correspond to standard deviations of at least three independent layer estimation) and d their surface exposure ratio as function of experimental time. e 3D model (not to scale) of the Pt3Ni growth process. The early stage model 1” is only constructed to show a possible dynamical cluster structure
In situ AP-XPS measurements
In situ AP-XPS experiments were conducted to characterize the elemental distribution near the particle surface region through the growth process by continuously collecting Pt 4f and Ni 2p signals under the synthesis condition (see experimental details in Methods section, Supplementary Fig. 10). Figure 2a, b shows the original Pt 4f and Ni 2p spectra collected according to the temperature profile in Fig. 2c. Peaks at 72.8 and 76.1 eV, corresponding to Pt2+ 4f7/2 and 4f5/217, and peaks at 855.7 and 873.1 eV, corresponding to Ni2+ 2p3/2 and 2p1/218, were identified prior to the experiments, indicating that both elements remained in their precursor state. A new peak emerged at 71.7 eV when the temperature was raised to around 120 °C. This new peak was assigned to Pt0 4f7/2, with its emergence suggesting an initiation of Pt reduction. Gradual growth of the Pt0 4f7/2 peak and diminishment of the Pt2+ 4f7/2 peak were observed with a further increase in the temperature, which indicated a continual Pt reduction process. Ni exhibited a delayed reduction compared to Pt, because the Ni0 2p3/2 peak at 852.7 eV did not appear until the temperature was above 150 °C. Control experiments were conducted with pure Pt(acac)2 and Ni(acac)2 to characterize their individual reduction processes (Supplementary Figs. 11, 12). The onset reduction temperature for Pt(acac)2 remained at around 120 °C, whereas Ni(acac)2 cannot be effectively reduced until above 200 °C. This interesting observation suggested that reduced Pt served as a catalyst for Ni reduction.

Surface composition evolution of the growing Pt3Ni nanoparticles. a Pt 4 f and b Ni 2p AP-XPS spectra and corresponding 2D projections of growing particles. c Temperature profile as a function of experiment time. d Calculated Pt0 and Ni0 contents near surface region of the growing particles based on quantitative XPS analysis (Error bars correspond to standard deviations of at least three independent composition analysis). e Growth model (not to scale) of Pt3Ni nanoparticle with near surface Pt0/Ni0 ratio changes (gray: Pt; purple: Ni)
Quantitative analysis of the AP-XPS data set (Supplementary Notes 3–5) yielded more detailed composition information of the growing particles in their near surface region (Fig. 2d), which provided an important basis for understanding the particle growth pathway. Because Pt reduction initiated at around 120 °C and no metallic Ni was detected until 150 °C, it could be concluded that Pt, rather than a Pt–Ni alloy, constituted the particles at their nucleation and early growth stage and served as seeds for the octahedral Pt3Ni formation. Above 150 °C, Ni began to be effectively reduced, as evidenced by a rapid increase in the Ni0 content. In addition, a gradual shift of Pt0 4f7/2 peak from 71.7 to 71.2 eV was observed in this stage (Fig. 2a), which can be attributed to electron transfer from Ni0 to Pt0 and was consistent with the quantitative surface composition change19. Interestingly, the near surface Ni0 content gradually reached nearly 50 at% at the late particle growth stage, which was significantly higher than its average content of 25 at% in the final Pt3Ni product. This discrepancy suggested the particles were Ni-enriched in the surface region, which was in good agreement with previous studies in literature20. Another interesting observation was that the surface Ni content, despite of its higher onset reduction temperature than Pt, reached 25 at% soon after its reduction and remained above this value until completion of the reduction process. It revealed, besides Ni reduction, that Ni in the bulk likely underwent a continual segregation to the surface region during the particle growth process. By combining the information obtained from STEM and AP-XPS, we were able to depict both morphological and surface composition changes of the growing particles. As illustrated in Fig. 2e, the growth of octahedral Pt3Ni starts with the formation of Pt nuclei and small spherical clusters, which continues to evolve through polyhedron to octahedron in morphology and evolve from pure Pt to about Pt50Ni50 in surface composition.
In situ XAS analysis
In situ XAS experiments were carried out to trace overall composition changes of the growing particles (Supplementary Notes 4, 5, Supplementary Fig. 13)21,22. Note that the experiments were conducted at a relatively lower temperature of 160 °C compared to that in AC-STEM and AP-XPS experiments so that the particle growth kinetics was sufficiently slow to allow for in situ XAS measurements, given that it took approximately 15 min to collect one XAS spectrum (see Methods section for experimental details). Our control experiments confirmed that such a change in reaction temperature altered the particle growth kinetics but would not affect the final particle morphology and composition (Supplementary Figs. 14, 15), which validated a combinational use of the in situ XAS data. Pt L3-edge and Ni K-edge spectra were collected in time series during the particle growth process (Fig. 3a, b and Supplementary Fig. 15). The extents of Pt and Ni reduction were evaluated based on changes in their XANES spectra. Figure 3c shows the plot of the percentage of remaining precursors at different reaction time. It was apparent that Ni reduction occurred more rapidly than Pt once it was initiated, as shown by a shorter period of time before the precursor depletion. The control XAS experiments with Ni(acac)2 showed little Ni reduction by itself under the same experimental condition (Supplementary Fig. 16). The results were in good agreement with the AP-XPS findings, which further supported our conclusion that Ni reduction was catalyzed by Pt. The completion of Ni reduction in a shorter time than that for Pt could lead to an argument that the grown particles should have Pt-enriched surfaces, which contradicted the AP-XPS observation of Ni enrichment in the surfaces. This contradiction can be resolved on the basis of Ni segregation to the surface during the octahedral Pt3Ni growth, as revealed by the AP-XPS data. This explanation was supported by post-treatment experiments with octahedral PtNi alloys in previous studies, which reported significant surface Ni segregation upon exposure to CO atmosphere at elevated temperatures23. Average coordination numbers for reduced Pt and Ni during the particle growth were calculated from the XAS data (Fig. 3d, Supplementary Table 1, Supplementary Fig. 17). The Pt–Pt coordination number remained higher than that of Pt–Ni throughout the reaction, and both gradually increasing with time. In comparison, the Ni-Pt coordination number was higher than that of Ni-Ni coordination and they seemed stabilized after the completion of the Ni reduction. The values of both ratios (Pt–Pt/Pt–Ni and Ni–Pt/Ni–Ni) in the octahedral Pt3Ni deviated from the Pt/Ni atomic ratio (i.e., 3:1) to a moderate extent. Considering the Pt enrichment in core region of the particles also contributed to such deviations, the results indicated that although Ni atoms were enriched on the surface, they were relatively well mixed with Pt atoms in the lattice.

Overall composition changes of the growing particles. a Pt L3-edge and b Ni K-edge XAS spectra of the growing nanoparticles. c Percentage of remaining precursors calculated from the XAS data and d coordination numbers of the reduced elements calculated from the XAS data as function of reaction time (Error bars correspond to fitting errors of XAS data processing)
First principle calculation of CO preferential adsorption
To understand the facet formation mechanism and the possible influences of Ni surface segregation on the facet formation, density functional theory (DFT) simulations were performed to substantiate our conjecture (Supplementary Fig. 18). This approach has already been used to effectively explain the morphology control mechanism under synthesis conditions5,24,25. The results showed that the enrichment of surface Ni significantly enhanced CO adsorption on the Pt3Ni (111) plane, which led to the formation of an octahedral particle (Fig. 4 and Supplementary Table 2). Compared with CO adsorption on Pt (111) (Eads = −2.04 eV), the adsorption of CO molecules was greatly strengthened at both Pt (−2.09 eV) and Ni (−2.23 eV) sites on Ni-enriched Pt3Ni (111). Although the Eads of CO at Pt site (−2.10 eV) on Ni-enriched Pt3Ni (001) was comparable with that at Pt site on the (111), the value at Ni site on the (001) facet (−2.16 eV) was less negative than that at the counterpart Ni site. The Eads of CO on Ni-enriched Pt3Ni (111) was about 0.05 eV more negative than that on the (001) on average. Although the energy difference was not dramatic, it altered the favored adsorption facet from the (001) planes for pure Pt to the (111) planes for Ni-enriched Pt3Ni that contributed to an octahedral morphology formation. Noticeably, DFT calculations showed that CO still preferred to adsorb on Pt3Ni (001) if there was no Ni enrichment at the surface (Supplementary Fig. 19), which further supported the crucial role of surface Ni enrichment in Pt3Ni for octahedral shape formation. Apart from the CO preferential adsorption, previous report indicates that the facet-to-facet atom diffusion would also play a role to shape the morphology owing to the different diffusion barriers26. Thus besides the observed dominant role of CO in controlling the octahedral Pt3Ni growth, it would be possible that surface atom diffusion on the growing particles also contributes to the facet formation.

CO adsorption energy on different facets. DFT calculation of CO adsorption energy on the (111) and (001) facets of surface Ni-enriched Pt3Ni
Discussion
A complete picture of the growth mechanism of octahedral Pt3Ni can be drawn by considering all of the information gained from the in situ experiments and DFT simulations. The STEM images with acceptable quality (identifiable lattice fringes and facets), except the early stage cluster, allowed us to build up the structure models. And XAS characterizations provided more detailed information like the atom distribution providing a clear picture of the growth pathway. Specifically, AP-XPS indicated a Pt-only stage, and both AP-XPS and XAS experiments confirmed the migration of Ni and Ni-rich-surface/Pt-rich-core properties. And the DFT calculations revealed the favorable CO adsorption on Ni-enriched Pt3Ni (111) facet. As shown in Fig. 5, Pt nuclei (early stage clusters) are first generated under the synthesis condition, which can be attributed to the facile reducibility of the Pt precursor. Ni then becomes catalytically reduced by Pt, leading to a mixed alloy formation. The continued reduction of Pt and Ni results in the growth in particle size and the evolution of low-index (111) and (001) facets. Along with the particle growth, Ni segregates outwards induced by CO and enriches the surface. CO molecules adsorb preferentially on the Ni-enriched (111) surfaces, which decreases their growth rate. This leads to the gradual growth of the (111) surfaces and diminishment of the (001) facets, and eventually to the formation of an octahedral morphology.

Schematic octahedral Pt3Ni growth pathway. Scheme (not to scale) of octahedral Pt3Ni growth pathway with inner section view (gray: Pt; purple: Ni). The nuclei stage is only constructed to show a possible dynamical structure
In conclusion, multiple cutting-edge in situ characterization techniques reveal the growth and facet formation pathway of octahedral Pt3Ni nanoparticles. Results indicate the surface enriched Ni plays a significant role in controlling the octahedra development by mediating the CO adsorption on (111) and (001) facets of Pt3Ni nanoparticles. Our findings highlight the significance of advanced in situ techniques in researching catalyst preparation and morphology control, providing an insightful understanding in synthesizing shaped catalysts.
Methods
Materials
Platinum acetylacetonate (Pt(acac)2, 97%) and nickel acetylacetonate (Ni(acac)2, 95%) were purchased from Sigma-Aldrich. Carbon black (C, Vulcan® XC-72R) was purchased from Cabot. Chloroform (CHCl3, 99.9%) was purchased from Fisher Scientific.
Sample preparation
The carbon supported Pt(acac)2 and Ni(acac)2 sample for in situ experiments was prepared via a dry impregnation method. In a typical procedure, 40 mg Pt(acac)2 and 17 mg Ni(acac)2 were dissolved in 3 mL chloroform, then the solution was added drop wisely onto 80 mg pretreated XC-72 carbon (300oC, overnight) under vigorous stirring. After the impregnation, the mixed sample (20 wt.% Pt loading, Pt:Ni = 3:1, molar ratio) was sealed in a glass vial and ready for the in situ characterizations. For comparison, the carbon supported pure Pt(acac)2 and pure Ni(acac)2 samples were also prepared through the same procedure.
In situ STEM
In situ scanning transmission electron microscopy (STEM) study of Pt3Ni octahedra growth was performed on a double Cs-corrected JEOL-JEM 3100-R05 microscope equipped with the Protochips AtmosphereTM gas cell system. In a typical method, the pre-mixed carbon supported precursor was dispersed in anhydrous methanol under sonication. Then the suspension was dropped directly onto a thermal E-chip and dried under the infrared lamp. The thermal E-chip is equipped with a thin ceramic heating membrane that can be controlled by the Protochips AtmosphereTM system. After that, a second E-chip window was then placed on top of the thermal chip in the holder, and this could create a thin gas chamber sealed from the high vacuum of the TEM column. The sample was situated between two 30–50 nm thick Si3N4 windows of the E-chips with a 5μm gap in between. The cross-section scheme of the assembled gas cell is shown in Supplementary Fig. 1. The operation accelerating voltage was 300 kV with a small probe current of 20pA, and the heating temperature for the gas cell was 200 °C (based on the Protochips calibration) with high purity H2/CO mixture gas (H2: 4%, CO: 96%, purity: 99.9995 %) flow at one atmosphere (760 Torr).
In situ AP-XPS
In situ AP-XPS experiments were performed at the 23-ID-2 (IOS) beamline of the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory. The end-station was equipped with a differentially pumped hemispherical analyzer (Specs Phoibos 150 NAP), which was offset by 70° from the incident synchrotron light and 20° from the surface normal of the sample (Supplementary Fig. 10). A more detailed description of the beamline and endstation can be found elsewhere27. A photon energy of 1100 eV was used for all experiments. The powder sample was pressed onto a tantalum foil, which was clipped onto a ceramic button heater. The temperature was monitored through a type K thermocouple placed underneath the foil. 100mTorr H2 and 500mTorr CO (Matheson, ultra-high purity) were introduced into the measurement chamber using variable leak valves, and the pressure was monitored using a capacitance manometer. The collected XPS spectra were calibrated based on the C1s = 284.5 eV as a reference. The Pt 4f and Ni 2p peaks were analyzed using a Shirley-type background with XPSpeak 4.1 software.
In situ XAS
X-ray absorption measurements were acquired on the bending magnet beamline of the Materials Research Collaborative Access Team (MRCAT) at the Advanced Photon Source, Argonne National Laboratory. Photon energies were selected using a water-cooled, double-crystal Si(111) monochromator, which was detuned by approximately 50% to reduce harmonic reflections. Measurements were made in transmission mode, and data points were acquired in three separate regions (energies relative to the elemental Ni K-edge 8333 eV and Pt L3-edge 11564 eV): a pre-edge region (−250 to −30 eV, step size = 10 eV, dwell time = 0.25 s), the XANES region (−30 to +30 eV, step size = 0.5 eV, dwell time = 0.25 s), and the EXAFS region (to 13 Å−1, step size = 0.07 Å−1, dwell time = 1 s). The ionization chambers were optimized for the maximum current with linear response (~1010 photons detected sec−1) 95% N2 and 5% Ar in the incident ion chamber and 40% N2 and 60% Ar in the transmission detector for both Ni edge and Pt edge measurement. A Ni or Pt foil spectrum was acquired simultaneously with each measurement for energy calibration. Samples were treated in a continuous-flow reactor, which consisted of a quartz tube (1-inch OD, 10-inch length) sealed with Kapton windows by two Ultra-Torr fittings (Supplementary Fig. 13). Ball valves were welded to each Ultra-Torr fitting and served as the gas inlet and outlet. An internal K type thermocouple (Omega) was placed against the sample holder to monitor temperature.
Samples (~10 mg) were pressed into a cylindrical sample holder consisting of six wells, forming a self-supporting wafer. The samples were treated at 160oC under H2/CO gas (2.5% H2, 50% CO and 47.5% Ar, ultra-high purity, 1 atm), and the XAS data were collected in situ.
XAS data processing
Standard procedures for normalization and background subtraction were performed using Demeter 0.9.25 software package. The edge energy of the X-ray absorption near edge structure (XANES) spectrum was determined from the inflection point in the leading edge, i.e., the maximum in the first derivative of the leading edge of the XANES spectrum. The pre-edge energies were also obtained in the first derivative using the zero-crossing point. The coordination parameters were obtained by a least square fit in R-space, k2-weighted Fourier transformed data using Artemis28.
DFT calculations
All DFT simulations were performed using the Quantum ESPRESSO package29. Structure relaxation and energy calculation were carried out through generalized gradient approximation (GGA) and the Perdew-Burke-Ernzerhof (PBE) functional with projector-augmented wave (PAW) sets from PSlibrary 0.3.130. The van der Waals (vdW) corrections were also included using the method of “Grimme-D2”31. The bulk lattice of Pt and intermetallic Pt3Ni were optimized prior to the cleavage of the (111) and (001) surface. The slab models were constructed using six-layer p(2 × 2) close-packed surface with a vacuum layer of 20 Å for both (111) and (001) surfaces. For the surface Ni enriched Pt3Ni model, we manually substituted the targeted Pt atoms in the upper three layers of Pt slab models and left the lower three layers Ni-free while keeping the overall stoichiometric ratio Pt:Ni = 3:1. For all the slab models, the bottom two layers are fixed during the structure relaxation. And for the CO adsorption, an on-top adsorption site was chosen to make the simulation consistent. We applied a plane-wave function cutoff of 50 Ry with a charge density cutoff of 500 Ry to initiate the calculations, and a k-point mesh of 5 × 5 × 1 was used. All the energies were calculated after the structures were fully relaxed. The adsorption energy for CO was calculated using the equation Eads = Esystem − Eslab − ECO. The weighted Eads for Pt and Ni co-exist surface was defined as Eads,w = Eads,Pt × θPt + Eads,Ni × θNi, where θPt and θNi are the surface coverage of Pt and Ni atoms in top layer.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Zhang, C., Hwang, S. Y., Trout, A. & Peng, Z. Solid-state chemistry-enabled scalable production of octahedral Pt–Ni alloy electrocatalyst for oxygen reduction reaction. J. Am. Chem. Soc. 136, 7805–7808 (2014).
Huang, X. et al. High-performance transition metal–doped Pt3Ni octahedra for oxygen reduction reaction. Science 348, 1230–1234 (2015).
Stamenkovic, V. R. et al. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 315, 493–497 (2007).
Choi, S.-I. et al. Synthesis and characterization of 9 nm Pt–Ni octahedra with a record high activity of 3.3 A/mgPt for the oxygen reduction reaction. Nano Lett. 13, 3420–3425 (2013).
Liao, H.-G. et al. Facet development during platinum nanocube growth. Science 345, 916–919 (2014).
Langille, M. R., Zhang, J., Personick, M. L., Li, S. & Mirkin, C. A. Stepwise evolution of spherical seeds into 20-fold twinned icosahedra. Science 337, 954–957 (2012).
Ievlev, A. V. et al. Quantitative description of crystal nucleation and growth from in situ liquid scanning transmission electron microscopy. ACS Nano 9, 11784–11791 (2015).
Li, Y. et al. In situ study on atomic mechanism of melting and freezing of single bismuth nanoparticles. Nat. Commun. 8, 14462 (2017).
Liao, H.-G., Niu, K. & Zheng, H. Observation of growth of metal nanoparticles. Chem. Commun. 49, 11720–11727 (2013).
Alloyeau, D. et al. Unravelling kinetic and thermodynamic effects on the growth of gold nanoplates by liquid transmission electron microscopy. Nano Lett. 15, 2574–2581 (2015).
Zheng, H. et al. Observation of single colloidal platinum nanocrystal growth trajectories. Science 324, 1309–1312 (2009).
Park, J. et al. 3D structure of individual nanocrystals in solution by electron microscopy. Science 349, 290–295 (2015).
Wu, J. et al. In situ environmental TEM in imaging gas and liquid phase chemical reactions for materials research. Adv. Mater. 28, 9686–9712 (2016).
Taheri, M. L. et al. Current status and future directions for in situ transmission electron microscopy. Ultramicroscopy 170, 86–95 (2016).
Shen, X. et al. In situ atomic-scale observation of the two-dimensional Co(OH)2 transition at atmospheric pressure. Chem. Mater. 29, 4572–4579 (2017).
Dai, S. et al. Revealing surface elemental composition and dynamic processes involved in facet-dependent oxidation of Pt3Co nanoparticles via in situ transmission electron microscopy. Nano Lett. 17, 4683–4688 (2017).
Xuyen, N. T. et al. Hydrolysis-induced immobilization of Pt(acac)2 on polyimide-based carbon nanofiber mat and formation of Pt nanoparticles. J. Mater. Chem. 19, 1283–1288 (2009).
de Souza, M. O., Mendes, F. M. T., de Souza, R. F. & dos Santos, J. H. Z. XPS characterization of nickel-acetylacetonate impregnated in NaX and NaY zeolites. Micro. Mesopor. Mater. 69, 217–221 (2004).
Nassr, A. B. A. A., Sinev, I., Pohl, M.-M., Grünert, W. & Bron, M. Rapid microwave-assisted polyol reduction for the preparation of highly active PtNi/CNT electrocatalysts for methanol oxidation. ACS Catal. 4, 2449–2462 (2014).
Cui, C., Gan, L., Heggen, M., Rudi, S. & Strasser, P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 12, 765 (2013).
Becknell, N. et al. Atomic structure of Pt3Ni nanoframe electrocatalysts by in situ X-ray absorption spectroscopy. J. Am. Chem. Soc. 137, 15817–15824 (2015).
Niu, Z. et al. Anisotropic phase segregation and migration of Pt in nanocrystals en route to nanoframe catalysts. Nat. Mater. 15, 1188 (2016).
Ahmadi, M., Cui, C., Mistry, H., Strasser, P. & Roldan Cuenya, B. Carbon monoxide-induced stability and atomic segregation phenomena in shape-selected octahedral PtNi nanoparticles. ACS Nano 9, 10686–10694 (2015).
Peng, Y.-K. et al. Mapping surface-modified titania nanoparticles with implications for activity and facet control. Nat. Commun. 8, 675 (2017).
Su, Y., Li, H., Ma, H., Robertson, J. & Nathan, A. Controlling surface termination and facet orientation in Cu2O nanoparticles for high photocatalytic activity: A combined experimental and density functional theory study. ACS Appl. Mater. Interfaces 9, 8100–8106 (2017).
Baletto, F., Mottet, C. & Ferrando, R. Molecular dynamics simulations of surface diffusion and growth on silver and gold clusters. Surf. Sci. 446, 31–45 (2000).
Palomino, R. M. et al. New in-situ and operando facilities for catalysis science at NSLS-II: the deployment of real-time, chemical, and structure-sensitive X-ray probes. Synchrotron Radiat. News 30, 30–37 (2017).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Paolo, G. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Dal Corso, A. Pseudopotentials periodic table: from H to Pu. Comput. Mater. Sci. 95, 337–350 (2014).
Stefan, G. Semiempirical GGA‐type density functional constructed with a long‐range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Acknowledgements
This work was supported by National Science Foundation (1665265) (Z.P.). S.Z., S.D., G.W.G., and X.P. acknowledge the National Science Foundation (grant numbers DMR-1506535 and CBET-1159240). G.Z. and J.T.M. would like to thank the National Science Foundation and the Center for Innovative and Strategic Transformation of Alkane Resources (CISTAR) for funding under Cooperative Agreement No. EEC-1647722. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Basic Energy Sciences [grant number DE-AC02-06CH11357]. MRCAT operations, beamline 10-BM, are supported by the Department of Energy and the MRCAT member institutions. This research used resources of the 23-ID-2 beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This research is also partially supported by Laboratory Directed Research and Development (LDRD15-037) program at Brookhaven National Laboratory.
Author information
Authors and Affiliations
Contributions
X.S., C.Z., S.Z., S.D., G.W.G., and Z.P. performed the in situ STEM characterizations. X.S., M.G., Y.P., A.H., and I.W. performed the in situ AP-XPS experiments. X.S., G.Z., and Y.P. conducted the in situ XAS tests. X.S., C.Z., Y.P., and S.M.S prepared the samples. X.S. performed the DFT simulations. X.S. analyzed the in situ STEM and AP-XPS data. X.S. and G.Z. analyzed the in situ XAS data. X.S. prepared the manuscript and I.W., J.T.M, X.P., and Z.P. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, X., Zhang, C., Zhang, S. et al. Deconvolution of octahedral Pt3Ni nanoparticle growth pathway from in situ characterizations. Nat Commun 9, 4485 (2018). https://doi.org/10.1038/s41467-018-06900-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06900-z
This article is cited by
-
Bifunctional interstitial phosphorous doping strategy boosts platinum-zinc alloy for efficient ammonia oxidation reaction and hydrogen evolution reaction
Nano Research (2024)
-
Mesoporous Pt@Pt-skin Pt3Ni core-shell framework nanowire electrocatalyst for efficient oxygen reduction
Nature Communications (2023)
-
Nanozymes based on octahedral platinum nanocrystals with {111} surface facets: glucose oxidase mimicking activity in electrochemical sensors
Microchimica Acta (2023)
-
In situ NMR reveals real-time nanocrystal growth evolution via monomer-attachment or particle-coalescence
Nature Communications (2021)
-
Gas adsorbate-induced Au atomic segregation and clustering from Cu(Au)
Science China Materials (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
