
Abstract
Innovative technologies are highly pursued for the detection and avoidance of counterfeiting in modern information society. Herein, we report the construction of photo-responsive supramolecular polymers toward fluorescent anti-counterfeit applications, by taking advantage of multicycle anthracene‒endoperoxide switching properties. Due to σ-metalation effect, photo-oxygenation of anthracene to endoperoxide is proceeded under the mild visible light irradiation conditions, while the backward conversion occurs spontaneously at room temperature. Supramolecular polymers are formed with cooperative nucleation‒elongation mechanism, which facilitate fluorescence resonance energy transfer process via two-component co-assembly strategy. Fluorescence resonance energy transfer efficiency is delicately regulated by either light-triggered anthracene‒endoperoxide conversion or vapor-induced monomer–polymer transition, leading to high-contrast fluorescent changes among three different states. On this basis, dual-mode anti-counterfeiting patterns have been successfully fabricated via inkjet printing techniques. Hence, cooperative supramolecular polymerization of photo-fluorochromic molecules represents an efficient approach toward high-performance anti-counterfeit materials with enhanced security reliability, fast response, and ease of operation.
Similar content being viewed by others
Introduction
Counterfeiting, widely spread in banknotes, diplomas, tax stamps, and certificates, has become a serious and social-threatening problem1. It is in keen pursuit to develop innovative anti-counterfeiting technologies, which avoids the forgeries and ensures security reliability for the genuine documents2,3,4,5. In this respect, photo-fluorochromic compounds are regarded as an intriguing choice, because of their reversible emission outputs in response to light6,7,8. Generally, inkjet printable anti-counterfeiting materials are prepared by direct mixing of photo-fluorochromic compounds with the polymer matrix9,10. However, their limited inclusiveness potentially leads to the decrease in ink’s quality. In addition, most of the reported anti-counterfeiting inks are operated in a single-mode fashion11,12,13,14,15,16,17, which can still be easily replicated. Hence, it is highly desirable to fabricate anti-counterfeiting inks with bright fluorescence and multi-mode responsiveness.
To attain this objective, a feasible approach is to graft photo-fluorochromic molecules onto polymer backbone11,12. An alternative strategy is to assemble such photo-responsive units into uniform supramolecular polymers18,19,20, which is superior to the covalent approach due to the avoidance of tedious synthesis. Moreover, supramolecular polymers serve as a dynamic platform for multi-component organization21,22,23,24,25,26,27, allowing for the cascade light-induced energy/electron transfer process28,29,30,31,32,33,34,35,36. As a consequence, the fluorescent signals can be fine-tuned in a multi-mode manner.
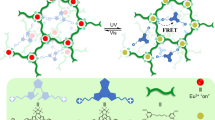
Based on these considerations, we sought to develop photo-fluorochromic supramolecular polymers toward anti-counterfeiting applications. The molecular design relies on anthracene‒endoperoxide interconversion, which displays on/off switching for the emission signals37,38,39,40,41. Notably, harsh reaction conditions are required for the traditional anthracene‒endoperoxide photo-switching (365 nm UV light irradiation for oxygenation process, while 254 nm UV light or thermolysis for deoxygenation reaction)37. Photo-degradation side-products are prone to form during the processes, which hamper the practical application in fluorescent anti-counterfeiting inks. To overcome this issue, herein anthracene-based monomer 1 (Fig. 1) has been designed, with the attachment of Pt(PEt3)2 moiety on both ends of 9,10-diacetylide anthracene. Due to σ-metalation effect42,43,44,45, photo-oxygenation of 1 can be rapidly proceeded via the less destructive visible light source, while the backward conversion occurs at room temperature in a spontaneous manner. These mild conditions facilitate the quantitative anthracene‒endoperoxide switching for multiple cycles. Additionally, two amide units are connected to the rod anthracene core of 1. Ascribed to the synergistic participation of hydrogen bonding and π‒π stacking interactions, supramolecular polymerization of 1 is expected to feature distinctive nucleation and elongation events46,47,48,49,50,51,52. It thus mimics the cooperative process adopted by biological self-assembled systems53,54, leading to macroscopic fluorescent variations before and after light irradiation. On this basis, a two-component supramolecular system 1/2 is established, which gives rise to fluorescence resonance energy transfer (FRET) from donor 1 to acceptor 2 (Fig. 1). The FRET efficiency can be precisely regulated, by either light-triggered anthracene‒endoperoxide or vapor-triggered monomer–polymer transitions. Accordingly, dual-mode anti-counterfeiting pattern (Fig. 1) in response to the orthogonal stimuli is achieved, which guarantees higher security reliability.

Molecular structures design and anti-counterfeiting applications. Schematic illustration of monomer 1 with anthracene‒endoperoxide photo-switching, and cartoon representation for the resulting supramolecular polymers toward fluorescent anti-counterfeiting applications
Results
Spectroscopic characterization of 1
As a first step, spectroscopic experiments were performed for the designed monomer 1. Specifically, it displays three main UV–Vis absorption bands in CHCl3 (Fig. 2a). According to the previous literatures55,56,57, the intense vibronic band at 250–310 nm, together with the moderately intense band at 400–525 nm (λmax = 454 and 484 nm for the two absorption maxima), are assigned to 1Lb and 1La bands of anthracene unit, respectively. In addition, the broad and intense band locating at 310–395 nm (λmax = 331 nm) belongs to π–π* transition of platinum acetylide (–C≡C–Pt–C≡C–) segment, which is absent for the counterpart compound 4 (see the structure in Fig. 2a, inset). Meanwhile, 1 shows the 1La emission band centered at 512 nm, along with a vibronic peak at 542 nm (Fig. 2b). As compared to 4, 1 displays ca. 50 nm bathochromic shifts for both 1La absorption and emission bands (Fig. 2a, b). Such phenomena are primarily ascribed to the overlapping between Pt d-orbitals and anthracene p-orbitals, resulting in the enhancement of π-electron delocalization45,55,56,57.

Spectroscopic characterization of 1. a UV–Vis and b fluorescence spectra of 1 and 4 in CHCl3 (2.00 × 10−4 M, λex = 425 nm for 1 and 408 nm for 4)
Supramolecular polymerization behaviors of 1
As widely documented, monomeric state dominates for π-conjugated molecules in dilute CHCl3 solution, while self-aggregation is more preferred in aliphatic media58. For 1 in methylcyclohexane (MCH), the 1La absorbance bands are centered at 451 and 480 nm at 298 K (Fig. 3a). It displays three isosbestic points (374, 460, and 487 nm) upon varying the temperature, suggesting the reversible transition between self-assembled and monomeric states (Fig. 3a). The strong aggregation tendency of 1 in aliphatic media can be further reflected by 1H NMR measurements. Briefly, broadened and unresolved aromatic resonances are present in d12-cyclohexane, which are in sharp contrast to the well-defined peaks observed in d-chloroform (Supplementary Fig. 2a). Moreover, the two amide units are prone to form intermolecular hydrogen bonds at room temperature, as evidenced by the upfield shifting of amide protons upon elevating the temperature (Supplementary Fig. 2b).

Characterization of supramolecular polymers from 1. a–b Temperature-dependent UV–Vis and CD spectra of 1 (2.00 × 10−4 M in MCH). Inset of b: thermo-responsive gel–sol transition of 1 (1.00 × 10−2 M in MCH). c Net helicity φn as a function of temperature for 1 (λ = 486 nm) and 3 (λ = 379 nm) (2.00 × 10−4 M in MCH). The red and pink lines denote the mathematical fitting. d TEM image by drop-casting 1 (4.00 × 10−4 M in MCH) on the copper grid. Scale bar: 500 nm
Circular dichroism (CD) spectroscopy is then utilized to probe the regularity of the supramolecular assemblies59. To induce the helical bias for 1 at the supramolecular level, optically active (S)-3,7-dimethyloctyl groups are introduced to the peripheral side chains (Fig. 1). At 298 K, no CD signals are detected for 1 in CHCl3 (Supplementary Fig. 1), because of the dominance of molecularly dissolved state. In sharp contrast, strong Cotton effects are observed for the 1La and 1Lb bands in MCH (for 448 nm, Δε = 33.8 L mol−1 cm−1, anisotropy factor g value = 0.0008; for 474 nm, Δε = 36.8 L mol−1 cm−1, g value = 0.00103; for 486 nm, Δε = –91.6 L mol−1 cm−1, g value = –0.0023, Fig. 3b). Meanwhile, no linear dichroism is detected for 1 (Supplementary Fig. 5), suggesting the measured CD signal is real to reflect the supramolecular chirality60. Accordingly, it supports the formation of long-range-ordered supramolecular polymers in apolar MCH medium, accompanying with the chirality transfer from the alkyl periphery to the inner anthracene unit59.
Supramolecular polymerization mechanism of 1 is further elucidated via temperature-dependent CD experiments. Specifically, CD signals completely lose upon elevating temperature to 322 K, and fully restore upon cooling (Fig. 3b). At the melting rate of 40 K h−1, no heating–cooling hysteresis can be observed, revealing that the self-assembly process is under thermodynamic control (Supplementary Fig. 3)18. When monitoring the CD intensity at 486 nm versus temperature, a non-sigmoidal melting curve is obtained for 1 (Fig. 3c). Hence, similar to the biological self-assembled polymers such as tobacco mosaic virus and cytoskeleton proteins53,54, 1 follows cooperative nucleation–elongation mechanism. For the counterpart compound 3 (see the structure in Fig. 1) with the replacement of anthracene unit on 1 by the benzene group, it also shows cooperative supramolecular polymerization behavior (Fig. 3c and Supplementary Fig. 4). Meijer–Schenning–van-der-Schoot mathematical model (Supplementary Equations 1 and 2) is employed to acquire the thermodynamic parameters for both self-assembly processes46,47. As shown in Table 1, monomer 1 features the higher Te, he and lower Ka values than those of 3, indicating the formation of more stable polymers with the higher cooperativity47. Considering that the structures between 1 and 3 only differ in the inner π-surface, it unambiguously supports that, in addition to intermolecular hydrogen bonds, π–π stacking interactions also participate in the supramolecular polymerization process.
Remarkably, 1 and 3 also display distinct gelation behaviors. In particular, when the monomer concentration of 1 exceeds 8 mM in MCH, it tends to form yellow transparent gels at room temperature (Inset of Fig. 3b). Depending on transmission electron microscopy (TEM) measurements, long ribbon-like fibers are observed for 1, with several microns in length and around 100 nm in width (Fig. 3d). These one-dimensional fibers are entangled to form fibrous networks, which lay the basis for the formation of supramolecular gels. In sharp contrast, neither gelation (even increasing the concentration to 80 mM) nor entangled long fibers can be visualized for 3 (Supplementary Figs. 6–7). Overall, minor modification on the monomeric structure brings about huge differences in supramolecular polymerization and macroscopic gelation behaviors.
Anthracene‒endoperoxide photo-switching of 1
We then turned to investigate the photo-responsiveness of 1. 460 nm LED lamp is employed, considering that 1 absorbs light in this region. Upon photo-irradiation of 1 in CHCl3, both 1La absorption (400–525 nm) and emission (475–650 nm) bands decrease rapidly in their intensities, and level off within 20 s (Fig. 4a and Supplementary Fig. 8). Two isosbestic points (318 and 354 nm) are observed in UV–Vis spectra, revealing the conversion from 1 to its photo-chemical product (Supplementary Fig. 8). The process can be further demonstrated by means of 1H NMR experiments (Fig. 5a, b), which show the disappearance of anthracene resonances (Ha 8.74 ppm, Hb 7.44 ppm), together with the emergence of a new set of resonances in the upfield region (Ha, 7.83 ppm, and Hb, 7.34 ppm).

Experimental and theoretical studies for photo-switching. a UV–Vis spectra change of 1 (2.00 × 10−4 M in CHCl3) upon the successive 460 nm light irradiation and standing at room temperature. Inset: multicycle tests by monitoring the absorbance changes at 484 nm. b Optimized structures and the Gibbs-free energy of 1 and 1-O2 on the basis of DFT calculations
The exact photo-chemical product is then elucidated. When N2 is purged into 1, both UV–Vis spectroscopic and 1H NMR signals hardly change (Supplementary Fig. 9). Accordingly, it suggests the participation of O2 during photo-conversion process. Furthermore, light irradiation is performed for the mixture of 1 and 2,2,6,6-tetramethylpiperidine (TEMP). In electron paramagnetic resonance spectroscopy, three narrow lines are observed with equal intensity (g value = 2.006)61,62, validating the formation of paramagnetic nitroxide radical, TEMPO (Supplementary Fig. 10). We rationalized that, due to Pt(II) σ-metalation effect, spin–orbit coupling takes place for 1 via intersystem crossing43. It thus facilitates the generation of singlet oxygen (1O2) via energy transfer between the triplet excited state of 1 and the surrounding O2, which is captured by TEMP. Hence, 1 undergoes rapid and quantitative [4 + 2] photo-oxygenation reaction with 1O2 to provide the endoperoxide species (1-O2, see Fig. 1). Density functional theory (DFT) calculations provide the extra evidence for [4 + 2] photo-transition process (Fig. 4b). In detail, the Gibbs-free energy (G) for 1, 1-O2, and 1O2 are calculated to be –4653.742376 a.u., –4803.871183 a.u., and –150.086995 a.u., respectively. The ΔG value for photo-oxygenation process is calculated to be –0.0418 a.u. (–109.7853 kJ mol−1), demonstrating the thermodynamic favorable conversion from 1 to 1-O2.
For endoperoxide-to-anthracene transition, deep UV light (254 nm) or thermolysis is commonly required for the conventional 9,10-disubstituted anthracenes37. In stark contrast, 1-O2 converts to 1 at room temperature in a spontaneous manner. The conclusion is verified by the reappearance of anthracene 1H NMR resonances (Fig. 5c), together with the complete restore of 1La and 1Lb spectroscopic bands (Supplementary Fig. 11). Half-life time (t1/2) for the deoxygenation process is determined to be 15.9 min by UV–Vis measurements (c = 2.00 × 10−4 M for 1-O2 at 293 K), which can be further modulated upon varying the temperature (3.70 min at 308 K, 8.60 min at 298 K, and 61.0 min at 288 K, see Supplementary Fig. 12). We rationalized that, when comparing to the anthracene unit in 1, the bulky endoperoxide ring in 1-O2 imposes larger hindrance to the neighboring two Pt(PEt3)2 units28. To release the steric stain, it could undergo spontaneous endoperoxide-to-anthracene transition for 1-O2 even at room temperature. With the successive light irradiation and room-temperature standing, 1 and 1-O2 can be reversibly switched for multiple cycles, which are confirmed by the vanishing/reappearance of both absorbance and emission bands (Inset of Fig. 4a, and Supplementary Fig. 11).

Anthracene‒endoperoxide photo-switching of 1. Partial 1H NMR spectra (CDCl3, 298 K, 2.00 mM) of 1: a before irradiation, b conversion to 1-O2 after 5 min irradiation, and c recovery to 1 upon room temperature standing for 10 min
We also performed anthracene‒endoperoxide photo-switching for the control compound 5 (see the structure in Fig. 6a, inset). Only 60% conversion takes place for the photo-oxygenation process, with the requirement of substantially longer irradiation time (800 s for 5 versus 20 s for 1 at 2.00 × 10−4 M, 298 K, see Fig. 6a and Supplementary Fig. 14). For the deoxygenation process (Fig. 6b), the t1/2 value for 5-O2 is determined to be 26 min, which is also slower than that of 1-O2 (8.6 min). Hence, it proves that Pt(II) σ-metalation of anthracene is essential for the rapid and quantitative anthracene‒endoperoxide switching.

Anthracene‒endoperoxide photo-switching kinetics. a Photo-oxygenation and b deoxygenation kinetics of 1 in CHCl3 (black square), in MCH (blue circle), and 5 in CHCl3 (red triangle) on the basis of UV–Vis measurements. For 1 and 5 (2.00 × 10−4 M, 293 K), the absorbance signals are monitored at 484 nm, and 470 nm, respectively
Photo-responsiveness of supramolecular polymers from 1
For 1, anthracene‒endoperoxide interconversion occurs not only in the monomeric state (CHCl3 solution), but also in the supramolecular polymeric state (MCH solution). Notably, the kinetics are different between the two states. In particular, the observed rate constant (kobs) for photo-oxygenation is 0.0128 s−1 in MCH, which is slower than that in CHCl3 (kobs = 0.71 s−1, see Fig. 6a and Supplementary Fig. 17). Similarity, for the backward conversion from 1-O2 to 1, t1/2 values vary from 15.9 min in CHCl3 to 34.0 min in MCH (Fig. 6b and Supplementary Fig. 18). The retarded conversion rates in supramolecular polymeric state are probably ascribed to the shielding of photo-fluorochromic units from 1O2. Another reason for the different kinetics originates from the varied dipolarity/polarizability for the two solvents63.
Photo-responsiveness of 1 gives rise to the macroscopic changes of the resulting supramolecular polymers. In detail, the bisignate CD signals of 1 in MCH totally disappear upon 460 nm light irradiation, denoting the loss of supramolecular chirality (Fig. 7a). Besides, supramolecular gels of 1 (10 mM in MCH) completely collapse within 4 min, accompanying with the weakening of green emission (Inset of Fig. 6b, and Supplementary Fig. 19). Upon standing at room temperature, highly emissive supramolecular gels reform, suggesting the complete deoxygenation of 1-O2.

Photo-switching of supramolecular polymers from 1. a CD and b fluorescent spectra of 1 (2.00 × 10−4 M in MCH at 298 K) upon 460 nm light irradiation for 260 s. Inset of b: gel–sol transition of 1 (10.0 mM in MCH) upon the successive 460 nm light irradiation and standing at room temperature
We then sought to exploit anti-counterfeiting applications of 1, by taking full advantage of its fluorescent on–off switching properties. Specifically, the supramolecular polymeric solution of 1 is filled in the customized cartridge of an inkjet printer. A monochromic QR code is printed on a non-fluorescent paper, which is invisible under natural light and can be decoded under UV light by a smartphone (Fig. 8a and Supplementary Fig. 21). When the printed paper is exposed to 460 nm light, the QR code is completely erased within 2 min. The initial state restores after standing at room temperature for 10 min, which is accelerated by heat treatment (3 min upon heating at 40 °C). Thanks to the quantitative anthracene‒endoperoxide photo-switching of 1, the inkjet-printed QR pattern can be read and hidden for multiple cycles (Fig. 8b).

Single-mode anti-counterfeiting inks from 1. a Photographs of the fluorescent QR code (size: 4 × 4 cm on paper) from 1 and its photo-responsiveness. Scale bar: 1 cm. b Relative fluorescence intensity (λ = 512 nm) for the QR code as a function of the cyclic numbers
Dual-mode anti-counterfeiting inks from copolymers 1/2
On this basis, FRET is introduced to the supramolecular polymeric system via a two-component co-assembly strategy (mixing monomers 1 and 2 together, see Fig. 1), which represents an effective route for the fabrication of multi-mode fluorescent anti-counterfeiting inks. For the efficient FRET process between 1 and 2, the prerequisite is their close distance and appropriate spectral overlap. Depending on the spectroscopic measurements, 2 features absorbance at 425–575 nm52, which overlaps well with the emission band of 1 (spectral overlap integral: J = 9.47 × 1011 M−1 cm−1 nm4, see Supplementary Figs. 22–23). Hence, monomers 1–2 are capable of serving as FRET donor–acceptor pairs.
The FRET efficiency is then evaluated, by progressive addition of 2 into the supramolecular polymeric solution of 1 (molar ratios of 1/2: from 500: 1 to 10:1). As shown in Fig. 9a, the emission signal of 1 (λ = 505 nm) decreases in its intensity, while that of 2 centered at 560 nm gradually increases, with an iso-emissive point at 541 nm. During the titration processes, the emission colors change from green to yellow (Inset of Fig. 9a, and Supplementary Fig. 24). For a 10:1 mixture of 1 and 2 in MCH, ΦET (energy transfer efficiency) and kET (energy transfer rate constant) are determined to be 80% and 2.40 × 109 s−1, respectively. Notably, neither emission spectra nor color changes can be observed for the 1/2 mixture in CHCl3 (Supplementary Fig. 25), suggesting that FRET occurs only in the co-assembled state.

Dual-mode anti-counterfeiting inks from 1/2. a Fluorescent spectra of 1 (4.00 × 10−5 M in MCH) via the gradual addition of 2 (0–10 mol%). b Fluorescent spectra of 1/2 (10 mol% of 2, 4.00 × 10−5 M for 1, λex = 425 nm) upon successive 460 nm light irradiation and room-temperature standing. Inset: intensity changes at 560 nm. c Dual-stimuli responsive dog pattern by inkjet printing 1/2 (4.00 × 10−5 M of 1 in MCH, 2.5 mol% of 2) on the weighting paper, together with the cartoon representation for the corresponding structures. The images are taken under 365 nm UV handhold lamp. Scale bars: 5 mm
A dog pattern with yellow emission is then obtained, by inkjet printing co-assembly 1/2 on the non-fluorescent papers (Fig. 9c and Supplementary Fig. 30). Upon 460 nm light irradiation, the emission signal is significantly weakened for its intensity (since 1-O2 is non-emissive, the weak fluorescence is derived from the trace amount of 2), while restored within 3 min at 40 °C. Considering that no chemical transformation takes place for 2 during the light irradiation processes (Supplementary Fig. 29), the fluorescent switching of 1/2 is unambiguously attributed to the interconversion between 1 and 1-O2, which deactivate/reactivate the FRET effect (Fig. 9b). In the meantime, fuming the dog pattern with CHCl3 vapor leads to the appearance of green emission, which promptly turns back to yellow upon drying (Fig. 9c). The phenomena are highly plausible, since polar solvent destroys the co-assembled structure and thereby shows the characteristic fluorescent signal of 1. More importantly, the anti-counterfeiting fluorescent inks derived from 1/2 feature sufficient stability and sustainability, since almost no emission decay can be visualized upon placing the printed dog pattern under the ambient conditions for 2 weeks (Supplementary Fig. 31). Overall, co-assembly 1/2 features three distinct emission signals (yellow, green, and weak fluorescence) upon light/vapor treatment, which is employed for dual-mode anti-counterfeiting applications with enhanced security reliability.
Discussion
In summary, we have successfully developed photo-responsive supramolecular polymers on the basis of multicycle anthracene‒endoperoxide switching. Due to Pt(II) σ-metalation effect, photo-oxygenation of 1 to 1-O2 can be proceeded via the mild visible light irradiation, while the backward conversion occurs spontaneously at room temperature. The fluorescent changes of 1 in response to light can be macroscopically amplified, because of the involvement of nucleation‒elongation cooperative supramolecular polymerization process. For the resulting supramolecular copolymers 1/2, FRET efficiency is orthogonally modulated via light and vapor stimuli, which facilitates the fabrication of dual-mode fluorescent anti-counterfeiting inks. Hence, supramolecular polymerization of photo-fluorochromic molecules provides an effective strategy toward high-performance anti-counterfeit materials with high security reliability, fast response, and ease of operation under the mild conditions.
Methods
Measurements
1H NMR spectra were collected on a Varian Unity INOVA-300 spectrometer with TMS as the internal standard. 13C NMR spectra were recorded on a Varian Unity INOVA-300 spectrometer at 75 MHz. MALDI-TOF measurements were recorded on a Bruker Autoflex Speed spectrometer with DCTB as the matrix. UV–Vis spectra were recorded on a UV-1800 Shimadzu spectrometer. CD measurements were performed on a Jasco J-1500 circular dichroism spectrometer, equipped with a PFD-425S/15 Peltier-type temperature controller. Solution excitation and steady-state fluorescence emission spectra were recorded on a FluoroMax-4 spectrofluorometer (Horiba Scientific) and analyzed with an Origin (v8.0) integrated software FluoroEssence (v2.2). TEM images were performed on a Tecnai G2 Spirit BioTWIN electron microscope (acceleration voltage: 120 kV). Electron paramagnetic resonance (EPR) measurements were performed on a JEOL JES-FA200 apparatus, with the utilization of 2,2,6,6-tetra-methylpiperidine as the spin trap for 1O2. Fluorescence microscopy images were performed on an inverted fluorescence microscope (Olympus IX81).
Theoretical calculations
All optimized structures were performed on Gaussian 09 software packages. All of the non-metallic elements (C, H, O, N, and P) were described by PBEPBE/6-31G computational method, while Pt atoms were described by LANL2DZ core potential. There are no imagery frequencies for the optimized geometries.
Inkjet printing experiments
Printing tests were performed on an HP inkjet office printer (HP Deskjet 2131 model) with the customized HP803 black ink cartridges. The filled inks (black) from the inkjet cartridge were removed, and the cartridge was washed extensively with ethanol, water, and dried with N2 blowing. The inks (0.5 mL) of 1 (1 mM) or 1/2 (2.5 mol% of 2, 0.5 mM for 1) were then loaded in the clean black ink cartridge to perform the printing experiments.
Data availability
The data that support the findings of this study are available from the corresponding author on request.
References
Berman, B. Strategies to detect and reduce counterfeiting activity. Bus. Horiz. 51, 191–199 (2008).
Yoon, B. et al. Recent functional material based approaches to prevent and detect counterfeiting. J. Mater. Chem. C 1, 2388–2403 (2013).
Andres, J., Hersch, R. D., Moser, J.-E. & Chauvin, A.-S. A new anti-counterfeiting feature relying on invisible luminescent full color images printed with lanthanide-based inks. Adv. Funct. Mater. 24, 5029–5036 (2014).
Jiang, K. et al. Triple-mode emission of carbon dots: applications for advanced anti-counterfeiting. Angew. Chem. Int. Ed. 55, 7231–7235 (2016).
Nam, H., Song, K., Ha, D. & Kim, T. Inkjet printing based mono-layered photonic crystal patterning for anti-counterfeiting structural colors. Sci. Rep. 6, 30885 (2016).
Yildiz, I., Deniz, E. & Raymo, F. M. Fluorescence modulation with photochromic switches in nanostructured constructs. Chem. Soc. Rev. 38, 1859–1867 (2009).
Andréasson, J. & Pischel, U. Smart molecules at work–mimicking advanced logic operations. Chem. Soc. Rev. 39, 174–188 (2010).
Irie, M., Fukaminato, T., Matsuda, K. & Kobatake, S. Photochromism of diarylethene molecules and crystals: memories, switches, and actuators. Chem. Rev. 114, 12174–12277 (2014).
Yoon, B. et al. An inkjet-printable microemulsion system for colorimetric polydiacetylene supramolecules on paper substrates. J. Mater. Chem. 22, 8680–8686 (2012).
Li, M. et al. Facile synthesis and screen printing of dual-mode luminescent NaYF4:Er,Yb (Tm)/carbon dots for anti-counterfeiting applications. J. Mater. Chem. C 5, 6512–6520 (2017).
Zhu, M.-Q. et al. Spiropyran-based photochromic polymer nanoparticles with optically switchable luminescence. J. Am. Chem. Soc. 128, 4303–4309 (2006).
Häckel, M., Kador, L., Kropp, D. & Schmidt, H.-W. Polymer blends with azobenzene-containing block copolymers as stable rewritable volume holographic media. Adv. Mater. 19, 227–231 (2007).
Klajn, R., Wesson, P. J., Bishop, K. J. M. & Grzybowski, B. A. Writing self-erasing images using metastable nanoparticle “inks”. Angew. Chem. Int. Ed. 48, 7035–7039 (2009).
Yoon, B. et al. Inkjet printing of conjugated polymer precursors on paper substrates for colorimetric sensing and flexible electrothermochromic display. Adv. Mater. 23, 5492–5497 (2011).
Qi, Q. et al. Solid-state photoinduced luminescence switch for advanced anticounterfeiting and super-resolution imaging applications. J. Am. Chem. Soc. 139, 16036–16039 (2017).
Wu, H., Chen, Y. & Liu, Y. Reversibly photoswitchable supramolecular assembly and its application as a photoerasable fluorescent ink. Adv. Mater. 29, 1605271 (2017).
Chen, C. et al. Facile synthesis of highly water-soluble lanthanide-Doped t-LaVO4 NPs for antifake ink and latent fingermark detection. Small 13, 1702305 (2017).
De Greef, T. F. A. et al. Supramolecular polymerization. Chem. Rev. 109, 5687–5754 (2009).
Yan, X., Wang, F., Zheng, B. & Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 41, 6042–6065 (2012).
Yang, L.-L., Tan, X.-X., Wang, Z.-Q. & Zhang, X. Supramolecular polymers: historical development, preparation, characterization, and functions. Chem. Rev. 115, 7196–7239 (2015).
Frisch, H. et al. pH-Switchable ampholytic supramolecular copolymers. Angew. Chem. Int. Ed. 52, 10097–10101 (2013).
Das, A. & Ghosh, S. Supramolecular assemblies by charge-transfer interactions between donor and acceptor chromophores. Angew. Chem. Int. Ed. 53, 2038–2054 (2014).
Chen, L.-J. et al. Hierarchical self-assembly of discrete organoplatinum(II) metallacycles with polysaccharide via electrostatic interactions and their application for heparin detection. J. Am. Chem. Soc. 137, 11725–11735 (2015).
Frisch, H. et al. Kinetically controlled sequential growth of surface-grafted chiral supramolecular copolymers. Angew. Chem. Int. Ed. 55, 7242–7246 (2016).
Ji, X. et al. Controlling amphiphilic copolymer self-assembly morphologies based on macrocycle/anion recognition and nucleotide-induced payload release. Chem. Sci. 7, 6006–6014 (2016).
Ji, X.-F., Xia, D.-Y., Yan, X.-Z., Wang, H. & Huang, F.-H. Supramolecular polymer materials based on crown ether and pillararene host-guest recognition motifs. Acta Polym. Sin. 9–18 (2017).
Wong, C.-H. et al. Formation of 1D infinite chains directed by metal–metal and/or π–π stacking interactions of water-soluble platinum(II) 2,6-bis(benzimidazol-2′-yl)pyridine double complex salts. J. Am. Chem. Soc. 140, 657–666 (2018).
Cardolaccia, T., Li, Y. & Schanze, K. S. Phosphorescent platinum acetylide organogelators. J. Am. Chem. Soc. 130, 2535–2545 (2008).
Ajayaghosh, A., Praveen, V. K., Vijayakumar, C. & George, S. J. Molecular wire encapsulated into π organogels: efficient supramolecular light-harvesting antennae with color-tunable emission. Angew. Chem. Int. Ed. 46, 6260–6265 (2007).
Abbel, R. et al. White-light emitting hydrogen-bonded supramolecular copolymers based on π-conjugated oligomers. J. Am. Chem. Soc. 131, 833–843 (2009).
Rao, K. V., Datta, K. K. R., Eswaramoorthy, M. & George, S. J. Highly pure solid-state white-light emission from solution-processable soft-hybrids. Adv. Mater. 25, 1713–1718 (2013).
Peng, H.-Q. et al. Biological applications of supramolecular assemblies designed for excitation energy transfer. Chem. Rev. 115, 7502–7542 (2015).
Liu, Y. et al. Protein-framed multi-porphyrin micelles for a hybrid natural-artificial light-harvesting nanosystem. Angew. Chem. Int. Ed. 55, 7952–7957 (2016).
Li, J.-J., Chen, Y., Yu, J., Cheng, N. & Liu, Y. A supramolecular artificial light-harvesting system with an ultrahigh antenna effect. Adv. Mater. 29, 1701905 (2017).
Huang, C.-B. et al. Real-time monitoring the dynamics of coordination-driven self-assembly by fluorescence-resonance energy transfer. J. Am. Chem. Soc. 139, 9459–9462 (2017).
Wang, H., Ji, X., Li, Z. & Huang, F. Fluorescent supramolecular polymeric materials. Adv. Mater. 29, 1606117 (2017).
Aubry, J.-M., Pierlot, C., Rigaudy, J. & Schmidt, R. Reversible binding of oxygen to aromatic compounds. Acc. Chem. Res. 36, 668–675 (2003).
Fudickar, W. & Linker, T. Why triple bonds protect acenes from oxidation and decomposition. J. Am. Chem. Soc. 134, 15071–15082 (2012).
Filatov, M. A. et al. Generation of triplet excited states via photoinduced electron transfer in meso-anthra-BODIPY: fluorogenic response toward singlet oxygen in solution and in vitro. J. Am. Chem. Soc. 139, 6282–6285 (2017).
Zhou, Y., Zhang, H.-Y., Zhang, Z.-Y. & Liu, Y. Tunable luminescent lanthanide supramolecular assembly based on photoreaction of anthracene. J. Am. Chem. Soc. 139, 7168–7171 (2017).
Liu, K., Lalancette, R. A. & Jäkle, F. B–N Lewis pair functionalization of anthracene: structural dynamics, optoelectronic properties, and O2 sensitization. J. Am. Chem. Soc. 139, 18170–18173 (2017).
Ko, C.-C. & Yam, V. W.-W. Transition metal complexes with photochromic ligands—photosensitization and photoswitchable properties. J. Mater. Chem. 20, 2063–2070 (2010).
Wong, W.-Y. & Ho, C.-L. Organometallic photovoltaics: a new and versatile approach for harvesting solar energy using conjugated polymetallaynes. Acc. Chem. Res. 43, 1246–1256 (2010).
Yam, V. W.-W., Au, V. K.-M. & Leung, S. Y.-L. Light-emitting self-assembled materials based on d8 and d10 transition metal complexes. Chem. Rev. 115, 7589–7728 (2015).
Ko, C.-C. & Yam, V. W.-W. Coordination compounds with photochromic ligands: ready tunability and visible light-sensitized photochromism. Acc. Chem. Res. 51, 149–159 (2018).
Jonkheijm, P., van der Schoot, P., Schenning, A. P. H. J. & Meijer, E. W. Probing the solvent-assisted nucleation pathway in chemical self-assembly. Science 313, 80–83 (2006).
Smulders, M. M. J., Schenning, A. P. H. J. & Meijer, E. W. Insight into the mechanisms of cooperative self-assembly: the “sergeants-and-soldiers” principle of chiral and achiral C3-symmetrical discotic triamides. J. Am. Chem. Soc. 130, 606–611 (2008).
Kaiser, T. E., Stepanenko, V. & Würthner, F. Fluorescent J-aggregates of core-substituted perylene bisimides: studies on structure−property relationship, nucleation−elongation mechanism, and sergeants-and-soldiers principle. J. Am. Chem. Soc. 131, 6719–6732 (2009).
Rest, C., Kandanelli, R. & Fernández, G. Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions. Chem. Soc. Rev. 44, 2543–2572 (2015).
Kulkarni, C. et al. Dipole-moment-driven cooperative supramolecular polymerization. J. Am. Chem. Soc. 137, 3924–3932 (2015).
Herkert, L., Sampedro, A. & Fernández, G. Cooperative self-assembly of discrete metal complexes. CrystEngComm 18, 8813–8822 (2016).
Wang, X. et al. Cooperative supramolecular polymerization of fluorescent platinum acetylides for optical waveguide applications. Angew. Chem. Int. Ed. 56, 12466–12470 (2017).
Oosawa, F. & Kasai, M. A theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 4, 10–21 (1962).
Klug, A. From macromolecules to biological assemblies. Angew. Chem. Int. Ed. Engl. 22, 565–582 (1983).
Nguyen, M.-H. & Yip, J. H. K. Gold (I) and platinum (II) tetracenes and tetracenyldiacetylides: structural and fluorescence color changes induced by σ-metalation. Organometallics 29, 2422–2429 (2010).
Nguyen, M.-H., Wong, C.-Y. & Yip, J. H. K. Ligand perturbations on fluorescence of dinuclear platinum complexes of 5, 12-diethynyltetracene: a spectroscopic and computational study. Organometallics 32, 1620–1629 (2013).
Li, Y., Köse, M. E. & Schanze, K. S. Intramolecular triplet energy transfer in anthracene-based platinum acetylide oligomers. J. Phys. Chem. B 117, 9025–9033 (2013).
Chen, Z., Lohr, A., Saha-Möller, C. R. & Würthner, F. Self-assembled π-stacks of functional dyes in solution: structural and thermodynamic features. Chem. Soc. Rev. 38, 564–584 (2009).
Lee, C. C., Grenier, C., Meijer, E. W. & Schenning, A. P. H. J. Preparation and characterization of helical self-assembled nanofibers. Chem. Soc. Rev. 38, 671–683 (2009).
Wolffs, M. et al. Macroscopic origin of circular dichroism effects by alignment of self-assembled fibers in solution. Angew. Chem. Int. Ed. 46, 8203–8205 (2007).
Moan, J. & Wold, E. Detection of singlet oxygen production by ESR. Nature 279, 450–451 (1979).
Liu, K. et al. Supramolecular photosensitizers with enhanced antibacterial efficiency. Angew. Chem. Int. Ed. 52, 8285–8289 (2013).
Aubry, J.-M., Mandard-Cazin, B., Rougee, M. & Bensasson, R. V. Kinetic studies of singlet oxygen [4+2]-cycloadditions with cyclic 1,3-dienes in 28 solvents. J. Am. Chem. Soc. 117, 9159–9164 (1995).
Acknowledgements
We acknowledge the funding support from National Natural Science Foundation of China (21871245 and 21674106), CAS Youth Innovation Promotion Association (2015365), and the Fundamental Research Funds for the Central Universities (WK3450000004).
Author information
Authors and Affiliations
Contributions
Z.G. and F.W. conceived the idea for this project. Z.G. performed the experiments, analyzed the data, and produced the artwork under the direction of F.W. Y.H. contributed to the theoretical calculations. All authors contributed to the manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, Z., Han, Y. & Wang, F. Cooperative supramolecular polymers with anthracene‒endoperoxide photo-switching for fluorescent anti-counterfeiting. Nat Commun 9, 3977 (2018). https://doi.org/10.1038/s41467-018-06392-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06392-x
This article is cited by
-
An all-in-one nanoprinting approach for the synthesis of a nanofilm library for unclonable anti-counterfeiting applications
Nature Nanotechnology (2023)
-
Cooperative supramolecular polymerization of styrylpyrenes for color-dependent circularly polarized luminescence and photocycloaddition
Nature Communications (2023)
-
Bioinspired thermadapt shape-memory polymer with light-induced reversible fluorescence for rewritable 2D/3D-encoding information carriers
Nature Communications (2023)
-
Cooperative Supramolecular Polymerization of Propeller-Shaped Triphenylamine Cyanostilbenes for Explosive Detection
Chinese Journal of Polymer Science (2023)
-
Development of highly sensitive metal-ion chemosensor and key-lock anticounterfeiting technology based on oxazolidine
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
