
Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) presents as the progressive fibrofatty replacement of the cardiomyocytes particularly in the right ventricular wall. Here, we report two cases with ARVC. In family A, the proband carries a Desmoglein2 (DSG2) gene complex heterozygous mutation NM_001943.4:c.146G>A/p.(Arg49His)and NM_001943.3:c.1592T>G/p.(Phe531Cys). In family B, the proband carries a homozygous mutation NM_001943.3:c.1592T>G/p.(Phe531Cys).
Similar content being viewed by others
Arrhythmogenic right ventricular cardiomyopathy (ARVC:OMIM#610193) is a fatal genetic cardiomyopathy with prevalence rate ~1/2000 to 1/50001. Cardinal manifestations typically involve right ventricular enlargement and dysfunction2. The disease is frequently inherited in an autosomal dominant mode with incomplete or complete penetrance, although autosomal recessive transmission has also been reported3. The main five cardiac desmosome components (desmoglein-2, DSG2; desmocollin-2, DSC2; desmoplakin, DSP; plakoglobin, JUP; plakophilin-2, PKP2) contribute to 50% or so of symptomatic individuals4,5,6,7.
Desmogleins are calcium-binding transmembrane glycoprotein components of desmosomes, which are cell–cell junctions between epithelial and myocardial8. The DSG2 gene encodes a key cadherin of the cardiac desmosome and the only desmoglein distributed in the cardiomyocytes. DSG2 is critical for the structural integrity of the intercalated discs, and a lack of DSG2-dependent adhesion is a major pathogenic mechanism of ARVC9,10. Recent studies have suggested that mutated DSG2 proteins are incorporated into desmosomes exhibit dominant-negative effects in ARVC11. In addition, mutations in DSG2 display a high degree of penetrance and result in varying levels of disease severity12,13. Moreover, patients with multiple desmosomal mutations have shown to have a severe clinical course with more ventricular arrhythmias and a higher frequency of heart failure than subjects with a single mutation14,15,16.
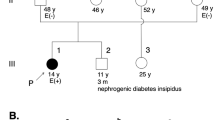
Family A: We experimented a male patient, a 7-year-old person (III-2 in Family A; Fig. 1a), presenting with abdominal distension and polypnea. He displayed corroborative evidence of heart failure, such as elevated levels of myocardial damage markers (N-terminal B-type natriuretic peptide [NT-BNP]: 4420 pg/mL, cardiac troponin [cTnI]: 0.046 μg/L). The 24 h dynamic electrocardiogram (DECG) revealed an incomplete right bundle branch block and epsilon waves (Supplemental Fig. S1). The echocardiogram showed enlarged right heart chambers (right atrial, RA = 59 × 57 mm; right ventricle, RV = 36 mm) (Supplemental Fig. S2). Cardiac magnetic resonance (CMR) confirmed RV abnormalities with focal bulges, excessive trabeculations localized in the RV apex. On the basis of these data, ARVC was diagnosed.

Pedigrees of the family A (a) and family B (e). Squares indicate males and circles females, ARVC patients are shown with solid symbols and unaffected with white symbols, carriers with white symbols plus solid point, the arrow indicates the proband. The proband’s (subject III:2) RNA sequence electropherograms are shown in (b). The DNA and amino aid sequences of DSG2 in family A (c, d) and family B (f)
The proband’s father (subject II:3) was first suspected to have ARVC at the age of 43 years when he exhibited an abnormal ECG with inverted T waves from V1 to V3 (Supplemental Fig. S3). His 24 h DCG showed premature ventricular contraction over 500/24 h (1151/24 h). The echocardiogram showed a mildly dilated RV (33 mm) (Supplemental Fig. S3). The proband’s mother (subject II:4) had no clinical symptoms at the age of 44 years and exhibited a normal ECG and echocardiogram.
Family B: We also experimented a male patient, a 29 years old (III-5 in Family B; Fig. 1e), who came to medical attention at the age of 23 years on account of recurrent palpitations, chest tightness. The electrocardiogram (ECG) was characterized by inverted T waves in V1–V4 (Supplemental Fig. S5). He exhibited increased right cardiac dimensions (RA = 48*42 mm, RV = 34 mm) with right ventricular aneurysm (11*7 mm) and broadening of the right ventricular outflow tract (RVOT = 38 mm) (Supplemental Fig. S5). CMR confirmed excessive trabeculations in the RV, thinner myocardium in the LV inferior wall and the LV apex, and mild LVEF reduction (49%). Becase of paroxysmal ventricular tachycardia, he was treated by radiofrequency catheter ablation (Supplemental Fig. S4). Based on the findings, the positive diagnosis of ARVC was made. When questioned regarding his family history, the patient mentioned that his grandfather (I-1: Fig. 1e) suffered from sudden cardiac death because of a stroke. His father (II-5: Fig. 1e) and his mother (II-6: Fig. 1e) shows no clinical symptoms and exhibited normal ECG and echocardiograms.
This study was approved by the Sichuan Academy of Medical Sciences and the Ethics Committee of the Sichuan Provincial People’s Hospital Trust. Informed consent were obtained from all individual participants included in this study. Clinical evaluation was based on the revised 2010 Task Force Criteria for ARVC.
Genomic DNA was extracted from peripheral blood samples with the Blood DNA Extraction Kit (Enriching Biotechnology, Shanghai, China). Briefly, using a customized Roche NimbleGen SeqCap EZ MedExome Kit (Roche Diagnostics, Wu Han, China), we targeted and enriched for the exons and the neighboring introns (within 50 bp) of the 130 genes associated with cardiomyopathy. Each quantified library was then loaded on the HiSeqXten platform (Illumina, Germany, Berlin) for next-generation sequencing. The detected variants were annotated and filtered using the following eight databases: ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), OMMI (https://www.ncbi.nlm.nih.gov/ommi/), RefGene (http://www.ncbi.nlm.nih.gov/RefSeq/), ExAC (http://exac.broadinstitute.org), Ensembl (http://www.ensembl.org), Encode (http://genome.ucsc.edu/ENCODE),1000 Genomes Project (http://www.1000genomes.org, 2014 Oct release), EVS (http://evs.gs.washington.edu/EVS). After filtering the candidates against multiple databases, the retained nonsynonymous single nucleotide variants were submitted to PolyPhen-2, SIFT, and Mutation Taster for functional prediction. Sanger sequencing was used to determine whether any of the remaining variants co-segregated with the disease phenotype in the two families. Sequencing data were compared to the Human Genome Database, and mutation naming followed the nomenclature recommended by the Human Genomic Variation Society (HGVS).
In the proband in family A, p.Arg49His was inherited from his father (Fig. 1b, c). The codon Arg49 is conserved among species (Fig. 2) and the p.Arg49His is predicted to be harmful by PolyPhen-2, SIFT, and MutationTaster. According to the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/450040/#summary-evidence), this variant is considered as “likely pathogenic” with the frequency of 0.000008 in ExAC. Previously, Awad et al.8 reported this variant as a compound heterozygous with the other variant. They predicted that the Arg49His would abolish furin cleavage of pro-desmoglein, thereby disrupting the production of mature, functional protein. Gandjbakhch et al.17 also described this variant as a consequence of de novo occurrence. Gaido et al.18 suggested that ARVC patients may exhibit extreme phenotypic-variavility in their clinical manifestations, even among patients carrying thep.Arg49His in the same family.

As determined using Human GRCh37/hg19, NM_001943.4:c.146G>A/p.(Arg49His) and NM_001943.3:c.1592T>G/p.(Phe531Cys) in DSG2 gene were hightly conserved across many species a. The DSG2 gene-related pathogenic or likely pathogenic missense mutations reported to date in patients with ARVC, the red ones indicate the mutations identified in the present study. Domains are exhibited with four green rectangles: Cadherin 1: Cadherin domain 165–262, Cadherin 2: Cadherin domain: 281–377, Cadherin 3: Cadherin domain: 400–490, Cadherin 4: Cadherin cytoplasmic region 778–841. The R49H variant at the head all of the domains, while F531C variant between Cadherin 3 and Cadherin 4 domain b
We consider that the p.Arg49His is a contributing factor to the severe phenotype of the proband in family A, in contrast with his father who carries the same variant; whereas the p.Phe531Cys inherited from his mother (Fig. 1b, d) would have aggravated the patient’s condition. Lin et al.19 reported the homozygous p.Phe531Cys in their cohort. This variant is likely to cause adverse changes in protein structure resulting in protein function changes that may ultimately weaken intercalated discs. Although the frequency of this variant in ExAC is reported as 0.00007, the recent study demonstrated that the p.Phe531Cys is highly prevalent (87%) among Chinese ARVC patients and has full penetrance for homozygous carriers20. This indicates that heterozygous carriers of this variant are unaffected.
In family B in this study, we identified p.Phe531Cys as a homozygous pattern (Fig. 1f). Based on the guideline for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, p.(Arg49His) and p.(Phe531Cys) were classified as likely pathogenic, respectively.
In conclusion, we identified homozygous and compound heterozygous variants with p.Arg49His in association with p.Phe531Cys a complex heterozygous mutation in family A, and careful follow-up of this family is necessary to elucidate whether the combinations of the variants are related to disease severity. Because of the limited cases of ARVC, further clarification of the proportion of patients with multiple mutations and their respective genotype–phenotype correlations remain to be explored.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2597
References
Corrado, D. et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 113, 1634–1637 (2006).
Basso, C. et al. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 9, 223–233 (2012).
Awad, M. M. et al. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, 258–267 (2008).
Corrado, D. et al. Arrhythmogenic cardiomyopathy. Circ. Res. 121, 784–802 (2017).
Rasmussen, T. B. et al. Mutated desmoglein-2 proteins are incorporated into desmosomes and exhibit dominant-negative effects in arrhythmogenic right ventricular cardiomyopathy. Hum. Mutat. 5, 697–705 (2013).
Zhou, X. et al. Comprehensive analysis of desmosomal gene mutations in Han Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Eur. J. Med. Genet. 58, 258–265 (2015).
Wada, Y. et al. Unique genetic background and outcome of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Mol. Genet. Genom. Med. 5, 639–651 (2017).
Awad, M. M. et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 79, 136–142 (2006).
Kant, S. et al. Desmoglein 2-dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ. Cardiovasc. Genet. 4, 553–563 (2015).
Vimalanathan, A. K. et al. Genetics of and pathogenic mechanisms in arrhythmogenic rightventricular cardiomyopathy. Biophys. Rev. 6, 973–982 (2018).
Dieding, M. et al. Arrhythmogenic cardiomyopathy related DSG2 mutations affect desmosomal cadherin binding kinetics. Sci. Rep. 7, 1–9 (2017).
Syrris, P. et al. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: agenotype-phenotype characterization of familial disease. Eur. Heart J. 28, 581–588 (2007).
Ohno, S. et al. Age-dependent clinical and genetic characteristics in Japanese patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. J. 77, 1534–1542 (2013).
Bauce, B. et al. Multiple mutations indesmosomal proteins encoding genes in arrhythmogenic rightventricularcardiomyopathy/dysplasia. Heart Rhythm. 7, 22–29 (2010).
Bhonsale, A. et al. Impact of genotype on clinical course in arrhythmogenic right ventriculardysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 14, 847–855 (2015).
Sen-Chowdhry, S. et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel Insightsinto patterns of disease expression. Circulation 13, 1710–1720 (2007).
Gandjbakhch, E. et al. Sporadic arrhythmogenic right ventricular cardiomyopathy/dysplasia due to a de novo mutation. Europace 11, 379–381 (2009).
Gaido, L. et al. Phenotypic expression of ARVC:How 12 lead ECG can predict left or right ventricle involvement. A familiar case series and a review of literature. Int. J. Cardiol. 236, 328–334 (2017).
Lin, Y. et al. Whole genome sequence identified DSG2 gene in a familial arrhythmogenic cardiomyopathy involving both ventricles. Cardiology 138, 41–54 (2017).
Chen, L. et al. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int. J. Cardiol. 274, 263–270 (2018).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81770379).
Author information
Authors and Affiliations
Contributions
X.C. collected the clinical data and drafted the manuscript. H.P. collected the clinical data and took regular follow-up, C.Z. analysed the sequencing data, H.Z. provided echocardiographic data, C.Y., H.M. and X.D. collected blood samples and send them for examination, X.L. participated in the design of the study and performed the statistical analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Peng, H., Zheng, C. et al. Two pedigrees with arrhythmogenic right ventricular cardiomyopathy linked with R49H and F531C mutation in DSG2. Hum Genome Var 6, 38 (2019). https://doi.org/10.1038/s41439-019-0069-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-019-0069-3
This article is cited by
-
Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy
Basic Research in Cardiology (2020)
