
Abstract
This study aimed to evaluate retinal structure in the early stage of Leber’s congenital amaurosis (LCA) caused by RPGRIP1 mutations. Four patients from two families were included. Case 1 was a 13-year-old girl, cases 2 and 3 were 7-year-old monozygotic twin brothers of case 1, and case 4 was a 17-year-old boy. Comprehensive ophthalmic examinations were performed, including visual acuity measurements, perimetry, electroretinography (ERG), and optical coherence tomography (OCT). To identify potential pathogenic mutations, 74 genes known to cause retinitis pigmentosa or LCA were assessed using targeted next-generation sequencing. OCT showed photoreceptor outer nuclear layer (ONL) thinning in all patients. The lamellar structure was retained in all patients, whereas the ellipsoid zone was extinguished in cases 1, 2, and 3. In case 4, the ellipsoid zone was maintained at 9 years of age but became blurred at 17 years of age. In case 1, OCT indicated slight photoreceptor ONL thinning during the period between 7 and 11 years of age. Mutation analysis revealed RPGRIP1 mutations as the cause for autosomal recessive LCA in all patients. Photoreceptor ONL on OCT is relatively well preserved in the early stage of LCA caused by RPGRIP1 mutations.
Similar content being viewed by others


Introduction
Leber’s congenital amaurosis (LCA) is one of the most severe forms of inherited retinal dystrophies. It is characterized by early-onset blindness or severe visual impairment during the first year of life, a specific behavior known as Franceschetti’s oculo-digital sign, congenital nystagmus, a sluggish pupillary reaction, and extinguished or severely reduced rod and/or cone responses on electroretinography (ERG)1. LCA is diagnosed primarily on the basis of severely impaired vision with some clinical characteristics in infancy, and genetic testing is known to play a role in confirmation of the diagnosis.
LCA is generally inherited as an autosomal recessive trait, although some families with autosomal dominant inheritance have been described2. The condition has been reported in many countries, and its worldwide incidence is 1–2 in 80,000 individuals3,4. Twenty-five causative genes have been identified5, including the RPGRIP1 gene, which encodes retinitis pigmentosa GTPase regulating interacting protein 1 and contributes to approximately 5% of cases of LCA6.
Several genotype–phenotype correlations that allow for the prediction of the gene from the phenotype characteristics have been confirmed. Koenekoop et al. described the correlation between gene defects and the natural history of visual function in patients with LCA. RPGRIP1 mutations are a degenerative type of mutation that are associated with a steady decline in visual function, while RPE65 mutations are an improvement type of mutation that are associated with a transient improvement and a subsequent decline in visual function7. The course of disease progression in LCA presents a challenge for gene therapy because the retention of sufficient photoreceptors is a prerequisite for a satisfactory therapeutic outcome. In an animal study, mice lacking RPGRIP1 showed highly disorganized photoreceptor outer segments at 20 days of age and photoreceptor loss by 5 months of age8. Accordingly, there is treatment potential for LCA associated with RPGRIP1 mutations. In a previous study in which gene therapy was performed on a murine model with RPGRIP1 mutations, ERG and histological examinations showed better preservation of photoreceptor function in the treated eyes9. In another study, gene therapy was effective in the early stage of disease in LCA patients with RPE65 mutations10. Although gene therapy can be more effective in patients in the early stage of LCA, to our knowledge, few studies have evaluated the retinal structure in LCA caused by RPGRIP1 mutations. In the present study, we evaluated the retinal structure in the early stage of LCA caused by RPGRIP1 mutations.
Materials and methods
Ethics statement
This retrospective study was approved by the Institutional Review Board of the Hamamatsu University School of Medicine (permit no. 14–040) and National Center for Child Health and Development (permit no. 686). The study protocol adhered to the principles of the Declaration of Helsinki. Written informed consent was obtained from the parents of all patients before any study procedure or examination was performed.
Clinical assessments
Four Japanese patients from two families who visited the National Center for Child Health and Development (NCCHD) were included in this study. These patients were diagnosed with LCA as in our previous study11. We reviewed the hospital records of all four patients, comprising the clinical findings and general medical and ophthalmic examinations, which included best-corrected visual acuity (BCVA) measurements, slit-lamp biomicroscopy, ophthalmoscopy after pupillary dilation, and Goldmann perimetry (GP) for kinetic visual field assessments. Fundus photography and fluorescein angiography (FA) were performed with the TRC-50LX (Topcon, Tokyo, Japan) and the RetCam imaging system (Natus Medical Inc., Pleasanton, CA, USA). The central retinal laminar architecture was evaluated using spectral-domain optical coherence tomography (SD-OCT; RS-3000, Nidek, Gamagori, Japan) and swept-source OCT (SS-OCT; Topcon). Full-field ERG was performed in accordance with the International Society for Clinical Electrophysiology of Vision protocol12. OCT and ERG were performed under general anesthesia.
Target capture and next-generation sequencing (NGS)
The integrity of the targeted NGS approach used in this study has been previously evaluated11. Library preparation for NGS was performed using the HaloPlex Target Enrichment Kit 500 kb (Agilent Technologies, Santa Clara, CA, USA) in accordance with the manufacturer’s instructions. A custom target enrichment library was designed to capture the 74 genes known to be associated with retinitis pigmentosa (RP) or LCA as reported in RetNet at the time of system design5. Probes were generated for 1182 regions to cover all exons and flanking intronic sequences (intronic sequence, ±25 bp from the exon boundaries) of the 74 genes. Amplicon libraries were prepared from the genomic DNA of patients in accordance with the manufacturer’s instructions13. A DNA sample library was quantified and loaded onto an MiSeq sequencer (Illumina, San Diego, CA, USA) in accordance with the manufacturer’s instructions using 150-bp paired-end chemistry.
NGS data analysis
All sequence and statistical analyses were performed using relevant programs in the commercially available, stand-alone Genomics Workbench software package (version 8.5.1; CLC bio, Aarhus, Denmark)13. We focused on nonsynonymous variants and splice site variants within 5 bp of the exon–intron boundaries (±5 bp) and excluded synonymous and noncoding exonic variants from the analysis. Common genetic variants (allele frequency, >0.005 for recessive variants or >0.001 for dominant variants) in any of the ethnic subgroups found in the following single nucleotide polymorphism (SNP) databases and synonymous variants were treated as possible nonpathogenic sequence alterations: 1000 Genomes database14, Exome Aggregation Consortium database15, Human Genetic Variation Database (HGVD)16, and Tohoku Medical Megabank Organization (ToMMo) database17. The HGVD and the ToMMo databases were used as a reference for Japanese controls. The Human Gene Mutation Database18 was used to screen for mutations reported in published studies.
Molecular validation of the candidate variants
Potential pathogenic mutations detected by NGS were validated using standard Sanger sequencing19. Sanger sequencing segregation analyses were performed for DNA from family members to investigate the cosegregation of potential pathogenic mutations. The following primer sets were used in the current study: exon 11 in the RPGRIP1 forward primer 5′-TGGGAAGATTAAATTCACACTTGA-3′ and reverse primer 5′-GTTAGTTTTCTAATCTCATCATCTTCC-3′ and exon 22 in the RPGRIP1 forward primer 5′-AAAGCAGTTGGTCCATGTTATTCT-3′ and reverse primer 5′- AGGTAATGGATTAGGTAGTCACAAA-3′.
Screening for the known exon 17 deletion mutation in RPGRIP111,20 was performed in the patient in case 4, in whom a targeted NGS approach had revealed a single heterozygous RPGRIP1 mutation. To identify the deletion breakpoints, long-range PCR and direct sequencing analysis were performed using the primers described by Suzuki et al.20
Results
Clinical findings
The clinical, visual field, and electrophysiological findings are summarized in Table 1.
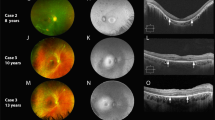
Case 1 (EYE20) was a 13-year-old girl who visited the NCCHD with congenital nystagmus and visual impairment at 7 years of age. Slit-lamp biomicroscopy showed normal findings. On funduscopic examination, both eyes showed retinal degeneration with attenuated vessels, chorioretinal atrophy, and peripheral pigmentation (Figs. 1, 2). At 7 years of age, full-field ERG showed a reduced rod response and an extinguished cone response (Fig. 3), while OCT showed a thinning photoreceptor outer nuclear layer (ONL) with a distinct adjacent lamellar architecture; notably, the ellipsoid zone was extinguished (Fig. 4a). At 11 years of age, ONL was not distinct in the perifoveal region, although lamellar structure was retained in the fovea (Fig. 4b). At 11 years of age, retinal aneurysmatic vessels with focal exudates (located in the inferior temporal mid-periphery) were treated with laser photocoagulation. Goldmann perimetry performed at 12 years of age revealed concentric constriction of the visual field. At 13 years of age, her decimal BCVA was 0.08 (+4.00/+3.00/5°) in the right eye and 0.1 (+4.00/+3.00/5°) in the left eye.

Fundus photographs of both eyes show attenuated retinal vessels (arrowheads) in cases 1–3 (EYE20, 64, and 65). In the photographs for case 4 (EYE55), the fundus lacks the typical changes seen in LCA

FA of both eyes showed granular hyperfluorescence without leakage due to retinal pigment epithelium atrophy in all four patients

The rod response is reduced (beginning at 7 years of age) in case 1 (EYE20). In cases 2 and 3 (EYE64 and EYE65, respectively), the rod response is abnormal (beginning at 1 year and 3 months of age). In case 4 (EYE55), the rod response is abnormal (beginning at 7 years of age). The cone responses are not detectable in all 4 cases

a The lamellar structure in the foveal area is retained in all four patients. In cases 1–3 (EYE20, 64 and 65), the ellipsoid zone is extinguished. In case 4 (EYE55), the ellipsoid zone is retained. Arrows indicate the lamina corresponding to the inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL) and ellipsoid zone (EZ). b OCT for case 1 (EYE20) shows changes in the macular structure with an increase in age. Although a continuous ONL with a distinct adjacent lamellar architecture is detectable in the foveal area on OCT performed at 7 years of age, ONL is not distinct in the perifoveal area (arrows) on OCT performed at 11 years of age. OCT indicates slight ONL thinning between 7 and 11 years of age. In OCT for case 4 (EYE55), the ellipsoid zone is maintained at 9 years of age but is blurred at 17 years of age (arrowheads)
Cases 2 and 3 (EYE64 and 65, respectively) were 7-year-old homozygotic twin boys diagnosed with LCA with congenital nystagmus and visual impairment at 1 year and 3 months of age. Slit-lamp biomicroscopy showed normal findings. On funduscopic examination, both eyes showed retinal degeneration with slightly attenuated vessels, chorioretinal atrophy, and mild peripheral pigmentation (Figs. 1, 2). Full-field ERG performed at 1 year and 3 months of age showed a subnormal rod response and an extinguished cone response (Fig. 3). OCT showed a thinning photoreceptor ONL with a distinct adjacent lamellar architecture at 1 year and 3 months of age; however, the ellipsoid zone was extinguished (Fig. 4a). At 7 years of age, BCVA was 0.06 (+4.00/+1.00/180°) in the right eye and 0.06 (+1.00/+3.00/180°) in the left eye for case 2, and 0.02 (+4.50) in the right eye and 0.02 (+5.50) in the left eye for case 3. Goldmann perimetry performed at 7 years of age showed concentric constriction of the visual field.
Case 4 (EYE55) was a 17-year-old boy diagnosed with LCA with congenital nystagmus and visual impairment at 7 years of age. Slit-lamp biomicroscopy showed normal findings. On funduscopic examination, both eyes appeared normal (Figs. 1, 2). Full-field ERG performed at 9 years of age revealed an extinguished cone response and a subnormal rod response (Fig. 3); OCT performed at 9 years of age showed a thinning photoreceptor ONL with a distinct adjacent lamellar architecture (Fig. 4a). The ellipsoid zone was maintained at 9 years of age but became blurred at 17 years of age (Fig. 4b). His BCVA at 15 years of age was 0.15 (−2.50/−1.50/°) in the right eye and 0.15 (−3.00/−1.50/°) in the left eye. Goldmann perimetry showed low sensitivity within each isopter.
Findings of targeted NGS and potential pathogenic mutations in the two families
These patients were diagnosed with LCA in our previous study11. Findings of targeted NGS are shown in the Supplementary Information and Supplementary Table 1. The obtained sequence data were analyzed using a previously described bioinformatics pipeline13. Cases 1–3 exhibited the homozygous deletion mutation c.3565_3571delCGAAGGC, whereas case 4 exhibited a heterozygous splicing mutation c.1467+1G>T and the heterozygous deletion mutation c.2710 + 374_2895 + 78del (Supplementary Figure, Supplementary Table 2, Supplementary Information)11.
Discussion
In the present study, we evaluated the retinal structure in four young patients with LCA caused by RPGRIP1 mutations. All patients in this study exhibited congenital nystagmus, poor visual acuity, and photophobia, which are characteristic of LCA. Cases 1–3 exhibited attenuated vessels, chorioretinal atrophy, and peripheral pigmentation; conversely, case 4 exhibited a normal fundus. Thus, these patients exhibited various, but not distinct, clinical findings compared with previously reported patients with RPGRIP1-associated LCA1,3,7,11,20,21,22. For example, although dysfunctions of both rod and cone photoreceptors are characteristic features of LCA, ERG on RPGRIP1-associated LCA is known to have a broader spectrum; Suzuki et al. described a patient who showed cone-dominant affected ERG20.
A few reports using OCT have mentioned that the central retinal laminar architecture, including the photoreceptor cell layer, is preserved in teenagers with RPGRIP1-associated LCA22. Wang et al. reported an abnormal macular structure identified using OCT at 5 years of age in a patient with RPGRIP1-associated LCA. Their OCT images demonstrated a relatively preserved foveal lamellar structure, decreased ONL thickness, and hardly visible ellipsoid zone band22. We evaluated the retinal structure on OCT in patients with RPGRIP1-associated LCA before adolescence; in particular, cases 2 and 3 were examined at 1 year and 3 months of age. To our knowledge, there have been no reports regarding OCT at such an early stage of RPGRIP1-associated LCA. The findings for cases 2 and 3 suggest that the ONL is better retained in early childhood than at 5 years of age22. In case 1, the ONL layer in the parafoveal area appeared thinner at 11 years of age compared with 7 years of age. In addition, in case 4, the ellipsoid zone was maintained at 9 years of age but became blurred at 17 years of age. These findings suggested that gene therapy in early childhood may provide better benefit to LCA patients with RPGRIP1 mutations.
RPGRIP1 is located on chromosome 14q11. It consists of 24 exons and encodes a protein with 1287 amino acids. The RPGRIP1 protein contains several structurally conserved motifs, a coiled coil (C2) domain, an RCC homology domain known as the RPGR interacting domain (RID), and a domain of unknown function23. RPGRIP1 interacts with RPGR in the cilium connecting the outer and inner photoreceptor segments, plays a role in ciliary trafficking in support of outer segment morphogenesis, and promotes outer segment development24. In addition, multiple proteins have synergistic actions; RPGRIP1 is necessary for RPGR binding to CEP29025, NPHP4 interacts with RPGRIP1 via a C2 domain26, and SPATA7 also interacts with RPGRIP1 via a C2 domain27. The loss of RPGRIP1 leads to the loss of RPGR in the connecting cilia. In a previous study, the loss of RPGRIP in mice resulted in a grossly oversized outer segment disc morphology8.
The homozygous deletion mutation c.3565_3571delCGAAGGC was detected in cases 1–3 in the present study. These patients exhibited phenotypic features similar to those already reported for RPGRIP1-associated LCA; a Korean patient with LCA who had the compound heterozygous deletion mutation c.3565_3571delCGAAGGC and missense mutation c.1892A>T showed photophobia and peripheral hyperpigmentation28. Case 4 exhibited a compound heterozygous splicing mutation c.1467+1G>T and an exon 17 deletion mutation. Suzuki et al. reported two Japanese brothers (7 and 11 years of age) with homozygous exon 17 deletion mutations in RPGRIP1; both were diagnosed with LCA and exhibited congenital nystagmus and visual impairment. They had a normal fundus and demonstrated recordable single-flash ERGs and undetectable 30-Hz flicker ERGs20. The c.1467+1G>T mutation was located in the intron 11 donor site. To predict the effect of the nucleotide substitution in the splice sites on splicing, we performed in silico analysis using splice site prediction29. The splice donor site score of the normal allele was 1.00, suggesting a high enough ability for splicing (score range of 0‒1 with a larger score indicating a greater ability), but the mutant allele was not recognized as a splicing donor site. Therefore, our results suggest that the c.1467+1G>T mutation causes intron 11 to remain in the transcript and undergo translation. This would produce a stop codon with 32 amino acids downstream, possibly resulting in nonsense-mediated mRNA decay (NMD)30. The splicing mutation c.1467+1G>T, resulting in a nonprotein product, and the lack of exon 17 would be a truncating mutation. Huang et al. suggested a correlation between the disease severity and the nature of the mutations in three patients with retinal dystrophy associated with RPGRIP1 mutations. The patient with the homozygous mutation ex1–22del was considered to have a severe form of the disease. The patient with the homozygous splicing mutation c.1468–2A>G, which is an in-frame mutation, was considered to have a less severe form of the disease, and the patient with the compound heterozygous nonsense mutation c.154C>T and missense mutation c.2020C>T was considered to have a less severe form of the disease31. According to that study, truncating mutations appeared to result in a more severe disease phenotype than did missense or in-frame splicing mutations. RPGRIP1 mutations result in retinal dystrophies with a broad range of phenotypes, ranging from LCA as a severe form to cone–rod dystrophy as a less severe form. In the present study, truncating mutations were detected in all four patients who exhibited characteristic LCA phenotypes.
This study is limited by the small number of patients. Further genetic analysis of a larger sample size and accumulation of clinical findings are necessary to understand the genotype–phenotype correlation in patients with RPGRIP1-associated LCA.
In conclusion, we described the features of young patients with LCA caused by RPGRIP1 mutations and evaluated retinal structure at a very early stage in patients with RPGRIP1-associated LCA. Although all four patients exhibited various clinical features, our findings suggest that the ONL on OCT is relatively well preserved in the early stage of RPGRIP1-associated LCA. Further analyses are needed to further clarify our findings.
References
Kumaran, N., Moore, A. T., Weleber, R. G. & Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 101, 1147–1154 (2017).
Bowne, S. J. et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 47, 34–42 (2006).
Stone, E. Leber congenital amaurosis—a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 (2007).
Koenekoop, R. K. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv. Ophthalmol. 49, 379–398 (2004).
Daiger S. D., Sullivan L. S. & Bowne S. J. The Retinal Information Network. (The University of Texas Health Science Center, Texas). http://www.sph.uth.tmc.edu/Retnet/. Accessed 23 Nov 2017.
Dryja, T. P. et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 68, 1295–1298 (2001).
Koenekoop, R. K., Lopez, I., den Hollander, A. I., Allikmets, R. & Cremers, F. P. Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions. Clin. Exp. Ophthalmol. 35, 473–485 (2007).
Zhao, Y. et al. The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc. Natl. Acad. Sci. USA 100, 3965–3970 (2003).
Pawlyk, B. S. et al. Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum. Gene Ther. 21, 993–1004 (2010).
Bainbridge, J. W. et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 372, 1887–1897 (2015).
Hosono, K. et al. Molecular diagnosis of 34 Japanese families with Leber congenital amaurosis using targeted next generation sequencing. Sci. Rep. 8, 8279 (2018).
Marmor, M. F. et al. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 118, 69–77 (2009).
Hosono, K. et al. Novel GUCY2D gene mutations in Japanese male twins with Leber congenital amaurosis. J. Ophthalmol. 2015, 693468 (2015).
The 1000 Genomes Project data. in The 1000 Genomes Project Consortium. http://www.1000genomes.org/. Accessed 13 Sep 2017.
ExAC database. in The Exome Aggregation Consortium. http://exac.broadinstitute.org/. Accessed 13 Sep 2017.
Higasa K., Miyake N. &, Yoshimura J. Human Genetic Variation Database. (Kyoto University, Kyoto). http://www.genome.med.kyoto-u.ac.jp/SnpDB/. Accessed13 Sep 2017.
Nagasaki, M., Yasuda, J., Katsuoka, F., Nariai, N., Kojima, K., Kawai, Y. et al. Rare variant discovery by deep whole–genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018 (2015).
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S. et al. Human Gene Mutation Database (HGMD). Hum. Mutat. 6, 577–581 (2003).
Hosono, K. et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE 7, e31036 (2012).
Suzuki, T. et al. A novel exon 17 deletion mutation of RPGRIP1 gene in two siblings with Leber congenital amaurosis. Jpn J. Ophthalmol. 58, 528–535 (2014).
Jacobson, S. G. et al. Leber congenital amaurosis caused by an RPGRIP1 mutation shows treatment potential. Ophthalmology 114, 895–898 (2007).
Wang, S., Zhang, Q., Zhang, X., Wang, Z. & Zhao, P. Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population. Graefes Arch. Clin. Exp. Ophthalmol. 254, 2227–2238 (2016).
Arts, H. H., Cremers, F. P., Knoers, N. V. & Roepman, R. Focus on molecules: RPGRIP1. Exp. Eye Res. 88, 332–333 (2009).
Won, J. et al. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum. Mol. Genet. 18, 4329–4339 (2009).
Gerner, M. et al. The retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1) links RPGR to the nephronophthisis protein network. Kidney Int. 77, 891–896 (2010).
Roepman, R. et al. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl Acad. Sci. USA 102, 18520–18525 (2005).
Eblimit, A. et al. Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Hum. Mol. Genet. 24, 1584–1601 (2015).
Seong, M. W. et al. Molecular characterization of Leber congenital amaurosis in Koreans. Mol. Vis. 14, 1429–1436 (2008).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 4, 311–323 (1997).
Hentze, M. W. & Kulozik, A. E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96, 307–310 (1999).
Huang, H. et al. Targeted next generation sequencing identified novel mutations in RPGRIP1 associated with both retinitis pigmentosa and Leber’s congenital amaurosis in unrelated Chinese patients. Oncotarget 8, 35176–35183 (2017).
Acknowledgements
The authors would like to thank Editage (https://www.editage.jp/) for English language editing. This work was supported by a grant from the Initiative on Rare and Undiagnosed Diseases for Adults (no. 16ek0109151h0002) from the Japan Agency for Medical Research and Development (AMED) and the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (no. 17K11447, awarded to Y.H., and no. 16K11284, awarded to K.H.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyamichi, D., Nishina, S., Hosono, K. et al. Retinal structure in Leber’s congenital amaurosis caused by RPGRIP1 mutations. Hum Genome Var 6, 32 (2019). https://doi.org/10.1038/s41439-019-0064-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-019-0064-8
