
Abstract
Ripe rot is a serious grapevine disease in Vitis L. and Muscadinia (Planch.) Small. However, resistance to this disease has been reported in some oriental Vitis species. To identify resistance-related Quantitative Trait Loci (QTLs) from the Chinese grape species V. amurensis, an F1 population of V. vinifera ‘Cabernet Sauvignon’ × V. amurensis ‘Shuang Hong’ was used to map the ripe rot resistance loci expected in ‘Shuang Hong’ grape. A total of 7598 single nucleotide polymorphisms (SNPs) between the parents were identified in our previous study, and 934 SNPs were selected for genetic map construction. These SNPs are distributed across the 19 chromosomes covering a total of 1665.31 cM in length, with an average of 1.81 cM between markers. Ripe rot resistance phenotypes among the hybrids were evaluated in vitro using excised leaves for three consecutive years from 2016 to 2018; a continuous variation was found among the F1 hybrids, and the Pearson correlation coefficients of the phenotypes scored in all three years were significant at the 0.01 level. Notably, the first QTL reported for resistance to grape ripe rot disease, named Cgr1, was identified on chromosome 14 of ‘Shuang Hong’ grapevine. Cgr1 could explain up to 19.90% of the phenotypic variance. In addition, a SNP named ‘np19345’ was identified as a molecular marker closely linked to the peak of Cgr1 and has the potential to be developed as a marker for the Cgr1 resistance haplotype.
Similar content being viewed by others


Introduction
Grape ripe rot disease, caused by Colletotrichum gloeosporioides (Penzig) Penz. & Sacc1. or Colletotrichum acutatum2, results in sunken necrotic lesions on stems, flowers, leaves, and fruit clusters3. In most grapevine planting regions of China, especially in southern China with rainy and humid veraison and maturity periods, C. gloeosporioides has become the main causal agent of grape ripe rot4.
Fungicide application is the most effective way to control grape ripe rot5. Because veraison and maturity are the main periods for C. gloeosporioides infection, application of fungicides is indispensable. The period between fungicide spraying and fruit ripening is too short to avoid the risk of introducing fungicide residues in berry and products derived from them (must, wine, and raisin). Garcia-Cazorla et al.6 detected fungicide residues in berry, must, and wine after monthly spraying of fungicides, and the amount of residues in berry was higher than those in must and wine. In addition, fungicide spraying is labor-intensive, costly, and damaging to the environment. Therefore, developing ripe rot-resistant varieties with high fruit quality would be beneficial to the grape industry.
Ripe rot, a serious disease in grapevine, has been reported in many species of Vitis L.4 and Muscadinia (Planch.) Small7. Li et al.8 evaluated ripe rot resistance in 56 accessions of Chinese wild Vitis species and found all of them to be resistant to ripe rot disease. Among them, there were 8 V. amurensis accessions, including ‘Shuang You’. ‘Shuang Hong’, which shares a common parent, ‘Shuang Qing’, with ‘Shuang You’, was used to investigate the genetics of ripe rot resistance in V. amurensis in this study.
Genetic mapping is commonly used for identifying genetic loci of interest. In grapevine, many genetic maps have been constructed using first- and second-generation markers, such as restricted fragment length polymorphisms (RFLP)9,10, amplified fragment length polymorphisms (AFLP)10,11, random amplification of polymorphic DNA (RAPD)9,11, and simple sequence repeats (SSR)12,13. However, the intervening distances among these markers are usually too long to fine-map the candidate genes. Single nucleotide polymorphisms (SNPs), as third-generation molecular markers, are the most abundant markers in grapevine and very useful for fine-mapping14,15,16. With the development of a library pooling strategy and high-throughput DNA sequencing technology17, SNP calling has developed into a time- and cost-saving marker technology. In the present study, we used a “genotyping-by-sequencing” strategy to identify SNPs for constructing genetic maps in a population of V. vinifera ‘Cabernet Sauvignon’ and V. amurensis ‘Shuang Hong’ to identify ripe rot resistance Quantitative Trait Loci (QTLs) in the latter.
Results
Construction of genetic map
The cross-pollination (CP) model of JoinMap 4.0 was used to construct the genetic maps. Markers were segregated into different linkage groups by the ‘independence LOD’ function, and a LOD score of ‘7’ was set as the threshold for determining whether loci were linked or not. Subsequently, the Kosambi function was used to calculate the distance between markers.
The map of the female parent, ‘CS’, contained 559 SNPs across 19 chromosomes (Chrs) covering 1506.57 cM, with an average marker distance of 2.93 cM (Table 1). The number of SNPs on the Chrs ranged from 13 (Chr09) to 43 (Chr18), with an average of 29.4 markers per Chr. The map of the male parent, ‘SH’, contained 511 SNPs covering 1440.14 cM across 19 Chrs, with an average marker interval of 3.36 cM (Table 1). The number of SNPs ranged from 12 (Chr18) to 42 (Chr13), with an average of 26.9 markers per Chr.
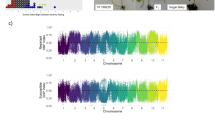
After construction of maps for the parents, the ‘Combine Groups for Map Integration’ function was used to construct the consensus map. This map contained 934 SNPs and covered 1665.31 cM on 19 Chrs. The average marker interval was 1.81 cM (Table 1; Fig. 1). The average number of SNPs per chromosome was 49.2. The genetic distance of the linkage groups ranged from 41.95 (Chr16) to 116.02 cM (Chr07), with an average length of 87.65 cM per Chr.

Integrated genetic map of ‘Cabernet Sauvignon’ × ‘Shuang Hong’
Phenotypic analysis of parents and progeny
Resistance to C. gloeosporioides was scored from 1 (most susceptible) to 9 (most resistant) (Fig. 2). The average scores for the parents during the three-year period (2016–2018) were 2.85 for CS and 7.86 for SH (Fig. 3). Resistance to C. gloeosporioides displayed continuous variation in the CS × SH hybrids and appeared to be a quantitative trait (Fig. 3). Most of the progeny fell into the resistance range of 6–7.99 (32.9–44.7%), followed by 8–9.00 (19.–35.3%). Some individuals showed transgressive segregation (Fig. 3). The Pearson correlation coefficient of the C. gloeosporioides resistance scores between 2016 and 2017 was 0.41, that between 2017 and 2018 was 0.40, and that between 2016 and 2018 was 0.28; all were significantly correlated at the 0.01 level (Table 2).

Leaf symptoms of ripe rot disease representing different levels of resistance/susceptibility

Distribution of ripe rot phenotype scores among 91 hybrids of ‘Cabernet Sauvignon’ × ‘Shuang Hong’
Nonparametric test
The rank-sum test, performed using the Kruskal–Wallis algorithm, identified 21 markers on Chr14 that cosegregated along with C. gloeosporioides resistance in three years (Table 3). Four of these 21 markers (np19345, np19481, np19678, and np19803) were identified in all three years, among which np19345 was the marker that cosegregated most significantly with C. gloeosporioides resistance.
QTL analysis
Based on the parent maps and the integrated map, interval mapping (IM) was used to identify the ripe rot resistance QTLs. In the map of ‘SH’ and the integrated map, a QTL on Chr14 was consistently observed in all three years evaluated. This QTL was named Cgr1. In 2016, the maximum LOD score (equal to 4.00) was located at 13.06 cM on Chr14 and explained 19.50% of the phenotype variance. The maximum LOD score was located at 12.06 cM in 2017 and 2018 and explained 16.00% and 17.20% of the phenotypic variance, respectively (Table 4, Fig. 4).

The dotted line indicates the LOD threshold
To fine-map Cgr1, np19345, the nearest marker to the two peak positions, was selected as the cofactor for multiple-QTL mapping (MQM) analysis. For the map of ‘SH’ and the integrated map, the same QTL could also be identified close to np19345 in all three years and explained up to 19.50% of the phenotypic variance (Table 4).
As expected, this QTL could not be identified in the female ‘CS’ map by either IM or MQM methods in any of the three years.
Correlation between C. gloeosporioides resistance and the np19345 marker
Np19345, the marker most significantly linked to C. gloeosporioides resistance as detected by the Kruskal–Wallis test (Table 3), was also the closest marker to the LOD peak in the Cgr1 region (Table 4). This marker was located at 4,080,914 bp on Chromosome 14 (Table S1). By analyzing the RAW sequencing data on this marker region, the nucleotides were ‘GG’ in ‘CS’ and ‘GA’ in ‘SH’, respectively (Fig. 5a). The progeny carrying ‘GG’ generally showed susceptible phenotypes, whereas ‘GA’ individuals generally showed resistance, and these results were quite consistent over all three years of disease evaluation (Fig. 5b–d).

a Base information of the np19345 allele and its flanking sequence; b–d Ripe rot phenotypes of F1 progeny with ‘GG’ and ‘GA’ in 2016 (b), 2017 (c), and 2018 (d)
Identification of putative ripe rot resistance genes
Seventeen biotic/abiotic stress-related genes were identified in the Cgr1 corresponding region from the grapevine reference genome ‘PN40024’ (Fig. 6). Among these, 11 genes were disease-related ‘R’ genes with NBS and/or LRR domains. Most of these disease-related genes were arranged in three clusters, which contained 3, 4, and 3 genes in regions of 94, 307, and 121 kb, respectively. Between clusters II and III, there was another NBS-LRR-like gene, one cell death-related gene, two frigida-like genes, and two superoxide dismutase [Cu–Zn] genes. Outside cluster III, there was EDR1, which was a general disease resistance- and stress tolerance-related gene.

Putative disease resistance genes identified in the grape ripe rot resistance QTL region based on physical locations from the grape reference genome ‘PN40024’
Discussion
Grape ripe rot caused by the fungal pathogen C. gloeosporioides is one of the most serious grapevine diseases. C. gloeosporioides can attack different parts of the grapevine tissues, but its main damage is to the ripening berries. The juvenile period of grape hybrids is variable; plants usually take 3-6 years to bear fruit, which makes it difficult to evaluate and select C. gloeosporioides resistance using berries in a timely fashion. To establish an in vitro evaluation system for ripe rot resistance in grapevine, Jang et al.18 inoculated ripe rot pathogens into several grapevine organs, including young leaves, mature leaves, young stems, and fruits, and they found that young leaves could be used for effectively predicting ripe rot resistance. In this study, we inoculated excised young leaves in three consecutive years and found that ripe rot symptoms appeared consistently. Based on the phenotype data, a QTL for ripe rot resistance was mapped on Chr14 of V. amurensis ‘Shuang Hong’. This result demonstrated that young leaves could be used for evaluating grape ripe rot resistance and mapping resistance QTLs.
The ripe rot resistances among the hybrids tended to increase from 2016 to 2018 (Fig. 3). This is likely because ripe rot resistance becomes stronger as the vine becomes older. This phenomenon also resulted in a narrower QTL region mapped by the IM method in 2018 than in 2016 and 2017 (Fig. 4).
To date, first-generation and second-generation molecular markers have been used to construct most grapevine genetic maps. In these maps, the average distance between markers mostly ranged from 4.6 cM to 11.5 cM19,20,21,22,23. Zyprian et al.24 constructed a linkage map with a combination of SSR and SNP markers, and the average distance between markers was reduced to 2.71 cM. Barba et al.16 used SNPs to construct two genetic maps, and the average distances between markers were 1.64 cM and 1.46 cM. In this study, SNPs were used to construct a genetic map with an average distance of 1.81 cM between markers. These results indicate that SNP markers are very frequent in grapevines and could significantly improve marker density compared to that of maps made using first- and second-generation markers.
Resistance (R) genes containing conserved domains, such as nucleotide-binding sites (NBS) and leucine-rich repeats (LRR), mainly function in disease resistance in plants25. In grapevine, at least 386 putative R genes have been predicted26, some of which play important roles in downy mildew27,28,29, powdery mildew30, and anthracnose29 resistance. Many of the disease resistance QTL regions were found to host a number of ‘R’ genes by comparison to the reference genome of the grapevine. For example, the corresponding region of Rpv10, a downy mildew resistance QTL identified on Chr9 from ‘Solaris’, contained 26 NBS-LRR genes corresponding to the genome region of ‘PN40024’13. The corresponding region of the Rpv1/Run1 locus, which cosegregated with both downy and powdery mildew, contained 11 NBS-LRR genes, and two of these 11 genes were verified as functional genes in response to the respective diseases31. In the present study, according to the ‘PN40024’ reference genome, the Cgr1 region contained 11 putative disease resistance genes with NBS and/or LRR domains (Fig. 6). Therefore, we believe that one or more of these predicted ‘R’ genes could determine ripe rot resistance in V. amurensis ‘Shuang Hong’ grapevine. In the future, we will interest to investigate them further to understand the genetic basis of resistance to grape ripe rot.
Materials and methods
Mapping population
The population was obtained by crossing V. vinifera ‘Cabernet Sauvignon’ and V. amurensis ‘Shuang Hong’ in 2011. Seeds (151) were germinated in a greenhouse in 2012, and seedlings (126) were planted in the greenhouse of the China Agricultural University experimental station, Haidian District, Beijing, China. The true hybrids (91) were confirmed using eight SSR markers (VMC1G3.2, VMC1G7, VMC5H5, VMC8G6, VRZAG83, VVIF52, VVIN56, and VVIP31).
Disease evaluation
The ripe rot pathogen, C. gloeosporioides, was isolated from an infected leaf and confirmed by sequencing C. gloeosporioides was cultured on potato dextrose agar (PDA) medium at 28 °C in the absence of light until the mycelium spread throughout the medium. After the mycelium was removed, the medium was cultured at 28 °C in the presence of light to promote spore production. The spore suspension was adjusted to ~100,000 spores per mL using sterile distilled water with 0.1% (v/v) Tween-20.
At the ten-leaf stage, which usually occurs in early May, sixteen square pieces (1-cm-side length) were sampled in each year from the fourth and fifth fully expanded leaves of each individual with surgical scissors and were placed on wet filter paper in Petri dishes with the abaxial side up. Leaf pieces were artificially inoculated by spraying with C. gloeosporioides suspension and stored in the dark at room temperature. Six days after inoculation, the disease symptoms were scored as 1, 3, 5, 7, or 9 based on the area of necrotic patches (1 = not limited, vast necrotic patches; 3 = numerous necrotic patches; 5 = limited necrotic patches; 7 = less necrotic patches; 9 = punctuated or no necrotic patches) (Fig. 2). The resistance score of each individual was equal to the average score of the 16 leaf pieces. Two leaf pieces from each of the progeny were inoculated with water as a control in each experiment.
DNA extraction
DNA was extracted from young leaves of the parents and progeny by the hexadecyl trimethyl ammonium bromide method, as described by Qu et al.32.
Genotyping and map construction
Library construction and sequencing were performed using the genotyping-by-sequencing method as described by Elshire et al.33 with minor modification. Mse I (New England Biolabs, Ipswich, MA, USA) and HaeII (New England Biolabs, Ipswich, MA, USA) were used to digest the DNA. Two adaptors with 6-nucleotide barcodes were ligated to the digested DNA fragments. By mixing all the samples together, we constructed a DNA pool. Primers complementary to the adaptor sequences were used to amplify the pool. PCR products between 400 and 425 bp were selected from agarose gel. Paired-end sequencing (PE150) was performed for the selected fragments using an Illumina 2500 platform (Illumina, San Diego, CA, USA) by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). Finally, we identified 7,598 high quality SNPs that could be used to construct a genetic map (Table S1) (unpublished). To avoid markers from the same genetic bin, the segregation type of each marker was analyzed, and only one SNP among markers of the same segregation type was retained. Moreover, we found that, if multiple-QTL mapping (MQM) with cofactor was selected for mapping by MapQTL 6.0 software, 50 markers was the threshold allowing computation in each linkage group for the cross CP population. Therefore, fewer than 50 markers on each chromosome that were distributed uniformly in physical distance were selected to construct the maps.
The CP model of JoinMap 4.0 was used to construct the linkage groups34. We selected lm × ll and hk × hk type markers to construct the map of the female parent (CS) and nn × np and hk × hk type markers to construct the map of the male parent (SH). Markers with too many missing genotypes or showing significantly distorted segregation (P < 0.001) were discarded. A LOD score equal to seven was the threshold to decide whether loci were linked. To optimize the marker order, markers with X2 > 3.0 were excluded. The Kosambi function was used to calculate the genetic distance between markers. After the parent maps were constructed, the ‘Combine Groups for Map Integration’ function was used to construct the integrated map.
QTL analysis
We used MapQTL 6.035 to calculate marker cosegregation and QTL position. The phenotypic (.qua file), map (.map file), and loci (.loc file) information were imported into MapQTL 6.0. The Kruskal–Wallis algorithm was employed as a nonparametric test to identify markers that were significantly associated with the trait. Interval mapping (IM) was used to detect putative QTLs related to the trait in a 0.5-cM step size. The marker close to the position with the highest LOD in each sampling year was selected as a cofactor. MQM was used for further accurate calculation of the putative QTLs detected by the IM test combined with the cofactor in 0.5-cM steps. The genomic-wide and LG-specific LOD threshold (α = 0.05) was calculated by 1000 permutation tests.
References
Southworth, E. A. Ripe rot of grapes and apples. J. Mycol. 6, 164–173 (1891).
Suzaki, K. Improved method to induce sporulation of Colletotrichum gloeosporioides, causal fungus of grape ripe rot. J. Gen. Plant Pathol. 77, 81–84 (2011).
Cannon, P., Damm, U., Johnston, P. & Weir, B. Colletotrichum–current status and future directions. Stud. Mycol. 73, 181–213 (2012).
Lin, T. et al. Differentiation in development of benzimidazole resistance in Colletotrichum gloeosporioides complex populations from strawberry and grape hosts. Austral. Plant Pathol. 45, 241–249 (2016).
Xu, X. et al. Characterization of baseline sensitivity and resistance risk of Colletotrichum gloeosporioides complex isolates from strawberry and grape to two demethylation-inhibitor fungicides, prochloraz and tebuconazole. Austral. Plant Pathol. 43, 605–613 (2014).
Garcia-Cazorla, J. & Xirau-Vayreda, M. Persistence of dicarboximidic fungicide residues in grapes, must, and wine. Am. J. Enol. Viticult. 45, 338–340 (1994).
Daykin, M. & Milholland, R. Histopathology of ripe rot caused by Colletotrichum gloeosporioides on muscadine grape. Phytopathology 74, 1339–1341 (1984).
Li, D., Wan, Y., Wang, Y. & He, P. Relatedness of resistance to anthracnose and to white rot in Chinese wild grapes. Vitis 47, 213–215 (2008).
Lodhi, M., Ye, G., Weeden, N., Reisch, B. & Daly, M. A molecular marker based linkage map of Vitis. Genome 38, 786–794 (1995).
Doligez, A. et al. Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor. Appl. Genet. 105, 780–795 (2002).
Dalbó, M. et al. A gene controlling sex in grapevines placed on a molecular marker-based genetic map. Genome 43, 333–340 (2000).
Goulão, L. & Oliveira, C. M. Molecular characterisation of cultivars of apple (Malus × domestica Borkh.) using microsatellite (SSR and ISSR) markers. Euphytica 122, 81–89 (2001).
Schwander, F. et al. Rpv10: a new locus from the Asian Vitis gene pool for pyramiding downy mildew resistance loci in grapevine. Theor. Appl. Genet. 124, 163–176 (2012).
Wang, N., Fang, L., Xin, H., Wang, L. & Li, S. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol. 12, 148–162 (2012).
Chen, J. et al. Construction of a high-density genetic map and QTLs mapping for sugars and acids in grape berries. BMC Plant Biol. 15, 28 (2015).
Barba, P. et al. Grapevine powdery mildew resistance and susceptibility loci identified on a high-resolution SNP map. Theor. Appl. Genet. 127, 73–84 (2014).
Yan, M. et al. Genotyping-by-sequencing application on diploid rose and a resulting high-density SNP-based consensus map. Hortic. Res. 5, 17 (2018).
Jang, M. H. et al. In vitro evaluation system for varietal resistance against ripe rot caused by Colletotrichum acutatum in grapevines. Hortic., Environ., Biotechnol. 52, 52–57 (2011).
Welter, L. J. et al. Genetic mapping and localization of quantitative trait loci affecting fungal disease resistance and leaf morphology in grapevine (Vitis vinifera L). Mol. Breed. 20, 359–374 (2007).
Zhang, J. et al. A framework map from grapevine V3125 (Vitis vinifera ‘Schiava grossa’ × ‘Riesling’) × rootstock cultivar ‘Börner’ (Vitis riparia × Vitis cinerea) to localize genetic determinants of phylloxera root resistance. Theor. Appl. Genet. 119, 1039–1051 (2009).
Blanc, S., Wiedemann-Merdinoglu, S., Dumas, V., Mestre, P. & Merdinoglu, D. A reference genetic map of Muscadinia rotundifolia and identification of Ren5, a new major locus for resistance to grapevine powdery mildew. Theor. Appl. Genet. 125, 1663–1675 (2012).
van Heerden, C. J., Burger, P., Vermeulen, A. & Prins, R. Detection of downy and powdery mildew resistance QTL in a ‘Regent’ × ‘RedGlobe’ population. Euphytica 200, 281–295 (2014).
Pap, D. et al. Identification of two novel powdery mildew resistance loci, Ren6 and Ren7, from the wild Chinese grape species Vitis piasezkii. BMC Plant Biol. 16, 170 (2016).
Zyprian, E. et al. Quantitative trait loci affecting pathogen resistance and ripening of grapevines. Mol. Genet. Genom. 291, 1573–1594 (2016).
McHale, L., Tan, X., Koehl, P. & Michelmore, R. W. Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212 (2006).
Goyal, N., Bhatia, G. & Singh, K. Identification of nucleotide binding site leucine rich repeats (NBS-LRR) genes associated with fungal resistance in Vitis vinifera. Preprint at https://pag.confex.com/pag/xxvi/meetingapp.cgi/Paper/27449 (2018).
Fan, J. et al. Characterization of a TIR-NBS-LRR gene associated with downy mildew resistance in grape. Genet. Mol. Res. 14, 7964–7975 (2015).
Kortekamp, A. et al. Identification, isolation and characterization of a CC-NBS-LRR candidate disease resistance gene family in grapevine. Mol. Breed. 22, 421–432 (2008).
Seehalak, W., Moonsom, S., Metheenukul, P. & Tantasawat, P. Isolation of resistance gene analogs from grapevine resistant and susceptible to downy mildew and anthracnose. Sci. Hortic. 128, 357–363 (2011).
Donald, T. et al. Identification of resistance gene analogs linked to a powdery mildew resistance locus in grapevine. Theor. Appl. Genet. 104, 610–618 (2002).
Feechan, A. et al. Genetic dissection of a TIR-NB-LRR locus from the wild north american grapevine species Muscadinia rotundifolia identifies paralogous genes conferring resistance to major fungal and oomycete pathogens in cultivated grapevine. Plant J. 76, 661–674 (2013).
Qu, X., Lu, J. & Lamikanra, O. Genetic diversity in muscadine and American bunch grapes based on randomly amplified polymorphic DNA (RAPD) analysis. J. Am. Soc. Hortic. Sci. 121, 1020–1023 (1996).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6, e19379 (2011).
Van Ooijen, J. JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. (Wageningen, Netherlands, 2006).
Van Ooijen, J. & Kyazma, B. MapQTL 6: software for the mapping of quantitative trait loci in experimental populations of diploid species. (Wageningen, The Netherlands, 2011).
Acknowledgements
This work was supported by the China Agriculture Research System (grant no. CARS-29-yc-2), the Start-up Fund from Shanghai Jiao Tong University (WF107115001) and Guangxi Bagui Scholar Fund (2013-3).
Author information
Authors and Affiliations
Contributions
J.L. conceived the study, designed the experiment, and helped to analyze the research results and prepare the manuscript. P.F. conducted the experiment, performed data analysis, and prepared the manuscript. Q.T., R.L., and G.L. assisted in the phenotyping experiments. S.S. helped with the linkage group construction.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41438_2019_148_MOESM1_ESM.xlsx
Table S1 Basic information of single nucleotide polymorphisms (SNPs) identified by the “genotyping-by-sequencing” method
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fu, P., Tian, Q., Lai, G. et al. Cgr1, a ripe rot resistance QTL in Vitis amurensis ‘Shuang Hong’ grapevine. Hortic Res 6, 67 (2019). https://doi.org/10.1038/s41438-019-0148-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-019-0148-0
This article is cited by
-
Construction of a resequencing-based high-density genetic map for grape using an interspecific population (Vitis amurensis × Vitis vinifera)
Horticulture, Environment, and Biotechnology (2022)
-
New quantitative trait locus (QTLs) and candidate genes associated with the grape berry color trait identified based on a high-density genetic map
BMC Plant Biology (2020)
-
Novel stable QTLs identification for berry quality traits based on high-density genetic linkage map construction in table grape
BMC Plant Biology (2020)
