
Abstract
In populations of facultatively sexual organisms, the proportion of sexually produced offspring contributed to each generation is a critical determinant of their evolutionary potential. However, estimating this parameter in natural populations has proved difficult. Here we develop a population genetic model for estimating the number of sexual events occurring per generation for facultatively sexual haploids possessing a biallelic mating-type locus (e.g., Chlamydomonas, ascomycete fungi). Our model treats the population as two subpopulations possessing opposite mating-type alleles, which exchange genes only when a sexual event takes place. Where mating types are equally abundant, we show that, for a neutral genetic marker, genetic differentiation between mating-type subpopulations is a simple function of the effective population size, the frequency of sexual reproduction, and the recombination fraction between the genetic marker and the mating-type locus. We employ simulations to examine the effects of linkage of markers to the mating-type locus, inequality of mating-type frequencies, mutation rate, and selection on this relationship. Finally, we apply our model to estimate the number of sexual reproduction events per generation in populations of four species of facultatively sexual ascomycete fungi, which have been jointly scored for mating type and a range of polymorphic molecular markers. Relative estimates are in line with expectations based on the known reproductive biology of these species.
Similar content being viewed by others


Introduction
The reproductive system of a species provides the genetic link between generations and has a profound influence on its evolutionary potential. Many species are facultatively sexual, contributing both asexual and sexually reproduced offspring to each generation. Asexual reproduction requires no interaction with mates and generates further copies of existing multilocus genotypes (de Meeus et al. 2007; Lopez-Villavicencio et al. 2013). In contrast, sexual reproduction via outcrossing with compatible partners re-assorts genetic variation, creating offspring with new allelic combinations, and breaks down non-random associations among alleles at different loci (Goddard et al. 2005; Nieuwenhuis and James 2016). Facultatively sexual populations practising a greater frequency of sexual outcrossing will be more burdened by the immediate ‘cost of sex’ (Lehtonen et al. 2012) but are anticipated to have a greater potential for long-term evolutionary responses to environmental change than their more asexual counterparts (Taylor et al. 2015). The latter will also tend to accumulate deleterious mutations that cannot be recombined away (Muller 1964; Goddard et al. 2005). Understanding the factors that influence the evolution of facultative sexual systems and testing hypotheses to account for variation in the numbers of mating types within a species requires quantitative measurements of s, the proportion of offspring produced by sexual outcrossing each generation (Billiard et al. 2012; Constable and Kokko 2018). Despite its critical importance, it has proved surprisingly difficult to obtain estimates of this parameter in natural and applied situations (Ali et al. 2016).
One approach for detecting deviations from full sexual reproduction in species such as fungi and protists that possess two distinct mating types is to determine the frequencies of the two mating-type ideotypes in snapshot samples (Linde et al. 2003; Siah et al. 2010). Under complete sexual outcrossing, a 1:1 ratio of ideotypes is anticipated, maintained by balancing selection (Milgroom 1996; May et al. 1999). Deviations from a 1:1 ratio of mating-type ideotypes should indicate a very low frequency or absence of sexual reproduction in the population. However, equal mating-type frequencies can be maintained by balancing selection even when significant asexual reproduction occurs, especially where population size is large and the effects of genetic drift are small (Milgroom 1996; May et al. 1999). Thus an analysis of mating-type frequencies alone generally provides limited information on the prevalence of sexual outcrossing in a species.
A second approach for investigating the extent of sexual outcrossing involves scoring a limited set of highly polymorphic molecular markers (10–20) and analysing the multilocus structure of populations (Maynard Smith et al. 1993; Milgroom 1996). If populations reproduce solely by sexual outcrossing, no linkage disequilibrium (LD) is expected between loci except where they are extremely tightly linked. However, where a proportion of offspring contributing to the next generation are produced by asexual reproduction, LD can be generated and maintained between unlinked or loosely linked loci by mutation, genetic sampling events (especially when populations are small) and by selection of particular genotypes (Hill and Robertson 1968; Maynard Smith et al. 1993). Thus the presence of LD between unlinked markers can potentially be an indication of departure from complete sexual outcrossing. However, empirical estimates of a standardised measure of LD (r2), derived from random samples, include a component of magnitude (1/sample size) that is due to sampling alone (Hill 1981; Waples 2006). This means that very large sample sizes (>200) are required to obtain accurate estimates of LD that are attributable solely to the presence of asexual reproduction. Furthermore LD can be generated by combining samples from populations that, individually, are completely sexual and at linkage equilibrium but differ in allele frequency. Thus it is not straightforward to use simple estimates of LD derived from a limited number of putatively unlinked markers to infer the extent of sexual reproduction in natural populations.
An alternative approach that employs LD to estimate the frequency of sex utilises mapped, high-density, single-nucleotide polymorphism (SNP) data that are now becoming available from resequencing studies. These can be used to calculate rates of decay of LD with map distance across the genome (Talas and McDonald 2015; Taylor et al. 2015; Nieuwenhuis and James 2016). Rates of decay of LD with map distance are expected to rise as rates of sexual outcrossing increase in the population, allowing rates of sexual reproduction to be compared across populations and species (Niewenhuis and James 2016). The simple relationship between sexual reproduction and decay of LD may be complicated by processes such as mitotic recombination and gene conversion (Hartfield et al. 2018). In the absence of these complications, the frequency of sexual reproduction can be calculated from data on the rate of decay of LD if the recombination rate across the relevant chromosome is available from laboratory studies (Tsai et al. 2008; Hartfield et al. 2018).
The final method for estimating the frequency of sexual reproduction in facultatively sexual species makes use of multilocus genotype data from individuals sampled from a known number of generations on either side of a sexual reproduction event (Ali et al. 2016). These data are used to determine the frequency with which individuals of the same clone (clonemates) are found both within and between generations from which the effective size of the population Ne and the frequency of sexual reproduction per generation s can be jointly estimated. Provided that clonemates can be detected at a reasonable frequency within and between generations, this is an elegant way of estimating s but relies on considerable background knowledge of the reproductive cycle of the species and the assumption that migration from outside the studied population is minimal (Ali et al. 2014, 2016).
Given the limitations of the analyses described above, there is considerable incentive for developing further population genetic methods for estimating rates of sexual outcrossing in facultatively sexual populations. Here we explore a new approach that is applicable to haploid populations in which sexual outcrossing is governed by two different ideotypes at a mating-type locus. Such populations are found widely in ascomycete fungi and protists, such as Chlamydomonas (Taylor et al. 2015; Nieuwenhuis and James 2016; Hadjivasiliou and Pomiankowski 2016). We describe a simple population genetic model that links the number of sexual outcrossing events per generation to a parameter FstM, which measures genetic differentiation caused by division of this population into two subpopulations possessing different ideotypes at the MT locus (Wright 1951). We use an analytical model to derive an expression for FstM under neutral processes and, using simulations, explore the behaviour of FstM when model assumptions are violated. On the basis of this information, we identify situations where the estimation of FstM can be useful for estimating the number of sexual outcross events per generation. The model is applied to population genetic data from four species of ascomycete fungi in which individuals have been scored simultaneously for mating type and a set of polymorphic molecular markers.
Population genetic model
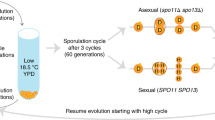
The life cycle and modes of reproduction of the haploid organisms considered here are illustrated in Fig. 1, where there is simultaneous sexual and asexual reproduction and a mixture of sexually and asexually reproduced individuals go forward to reproduce the population in each generation. The population genetic model describes the behaviour of a population of effective size Ne that is polymorphic at the mating-type locus MT with two ideotypes (MT-1 and MT-2), denoted by alleles M and m, with frequencies p and (1−p), respectively. The reproductive population, contributing offspring in each generation, can be thought of as two subpopulations, one with Nep individuals possessing the MT-1 ideotype, and the other with Ne(1−p) individuals possessing the MT-2 ideotype. Within this reproductive population, sexual reproduction occurs only between different mating types and contributes a proportion s of offspring to each mating-type subpopulation. Asexual reproduction occurs within each mating type and contributes offspring to each mating-type subpopulation with a probability (1−s).

The life cycle and modes of reproduction of the haploid organisms considered in the model. Sexually produced individuals are shown as solid, asexually reproduced as hatched. FstM measures genetic differentiation between the mating-type subpopulations
Consider a diallelic neutral polymorphic locus A with alleles A and a at frequencies q and (1−q), respectively, in the total population. The recombination rate between locus A and MT is r. Sexual reproduction provides the opportunity for gene flow to occur between the two mating-type subpopulations. If sexual reproduction occurs, the probability of gene migration at locus A between the two mating-type subpopulations is given by r. Let u be the mutation rate from A to a per generation and v be the mutation rate from a to A per generation. We assume that the demography of the fungal population is stable, with a constant frequency of each mating type per generation, denoted by \(\widehat p\left( {0 < \widehat p < 1} \right)\) for M. Let q1 and q2 be the frequencies of allele A conditional on the mating type M and m chromosomes, respectively. Thus \(q{\mathrm{ = }}\widehat pq_1 + \left( {1 - \widehat p} \right)q_2\). According to Bayes formula, the gametic frequencies in the total population can be expressed as \(p_{MA} = \widehat pq_1\), \(p_{ma} = \left( {1 - \widehat p} \right)\left( {1 - q_2} \right)\), \(p_{Ma}{\mathrm{ = }}\hat p\left( {1 - q_1} \right)\), and \(p_{mA} = \left( {1 - \widehat p} \right)q_2\). LD, denoted by D, between loci A and MT in the total population is expressed as
Under the joint effects of genetic drift, mutation and sexual reproduction (equivalent to migration), a steady-state distribution of genetic variation in the whole population or within and between subpopulations at locus A is eventually attained. The gene diversity (analogous to average heterozygosity in a diploid case) at locus A in the subpopulation with mating type MT-1, denoted by hM, is \(h_M = 1 - q_1^2 - \left( {1 - q_1} \right)^2\). This measure is re-expressed as \(h_M = 2q\left( {1 - q} \right) - 2D\left( {2q - 1} \right)/\widehat p - 2D^2{\mathrm{/}}\widehat p^2\) since \(q_1 = q + D/\widehat p\). Similarly, the gene diversity at locus A in the subpopulation with mating type MT-2, denoted by hm, is \(h_m = 1 - q_2^2 - \left( {1 - q_2} \right)^2\), which can be re-expressed as \(h_m = 2q\left( {1 - q} \right) + 2D\left( {2q - 1} \right)/\left( {1 - \widehat p} \right) - 2D^2/\left( {1 - \widehat p} \right)^2\). The average gene diversity (heterozygosity) within subpopulations at locus A \(\left( { = \widehat ph_M + \left( {1 - \widehat p} \right)h_m} \right)\) is \(2q\left( {1 - q} \right) - 2D^2{\mathrm{/}}\widehat p\left( {1 - \widehat p} \right)\). The expected heterozygosity in the total population is \(2q\left( {1 - q} \right)\). Thus, according to Wright (1969, pp. 294–295), genetic differentiation between the two subpopulations, measured by FstM, is given by
which has the same form as the square of the standardised LD except that \(\hat p\) is assumed to have a known prior value.
According to Ohta and Kimura (1969), the stationary distribution of any function (f) of variables q and D satisfies Kolmogorov backward equations for multiple variables. We can use this approach as a means of incorporating the effects of mutation and genetic drift into our model. In Supplementary Materials, we use reasoning similar to that of Ohta and Kimura (1970) to derive the following equation:
The E in Eq. (3) stands for the expectation with respect to the stationary distribution of function f. Note that Eq. (3) has both different velocities and diffusion coefficients from those in Eq. (9) of Ohta and Kimura (1970).
Letting f = D in Eq. (3) yields E(D) = 0. Thus, under the neutral process, no LD is expected at the steady state. Letting f = q in Eq. (3) yields \(E\left( q \right) = \nu {\mathrm{/}}\left( {\mu + \nu } \right)\), indicating that the expected allele frequency in the total population is not affected by the sexual reproduction process. Substitutions of f in Eq. (3) with D2, q2 and qD can, respectively, yield different equations for E(D2), E(q2) and E(qD). Using these equations in conjunction with Eq. (2) and with application of the approximation E(X/Y) ≈ E(X)/E(Y) (see Supplementary Materials for full details), we can derive the following expression involving FstM:
In the case of equal subpopulation sizes (\(\widehat p\) = 1/2), the value of FstM is maximised so long as more than one individual (Nesr > 1) per generation is involved in sexual reproduction. Under equal subpopulation sizes, Eq. (4) simplifies to
Where the rate of migration of alleles between mating-type subpopulations of equal size is much greater than the mutation rate, i.e. \(sr > > \left( {\mu + \nu } \right)\) and FstM is measured using markers unlinked to the mating-type locus (r = 0.5), the number of sexual mating events per generation Nes can be estimated as:
Using the delta method yields an approximation of the variance V(Nes) as
from which the standard deviation of the estimate of Nes can be derived. This approximation is appropriate when sample size is reasonably large, say >30 individuals.
Simulation modelling
Aims
To evaluate the effectiveness of expression (6) in relating genetic differentiation between the two mating-type populations (FstM) to the number of sexual events per generation (Nes), we conducted simulations of the process and compared our simulation results with the theoretical predictions developed above under different scenarios. The aims were to look at the effects, on FstM and the standard errors of FstM and LD, of varying the following parameters: the frequency of sexual reproduction per generation (s), the recombination rate (r) between the A and MT loci, the effective population size (Ne), the relative frequencies of the two mating types (p), the mutation rates per generation (µ and v), and various forms of selection. Note that p was held constant at p = 1/2 in simulating the effects of all factors except the effect of variation in p itself.
Simulation procedure
Scripts used in the simulations are provided in the Data Archive (three programs). The simulated samples were generated using the following procedure. We initially set the effective population size (Ne), the proportion of individuals with the MT-1 ideotype (p), the frequencies of alleles A and a conditional on chromosomes carrying alleles M (q1 and 1−q1) and m (q2 and 1−q2) and the recombination rate r between the MT and A loci. The initial haplotype frequencies for all simulations were set as q1 = 1 for allele A in the MT-1 subpopulation and q2 = 0 for allele A in the MT-2 subpopulation. Allele frequencies were calculated after joint asexual (rate (1−s)) and sexual (rate s) reproduction. A recurrent mutation process was introduced at the A locus based on a Poisson distribution for the number of mutants (Neµ and Nev per generation), and the conditional allele frequencies (q1 and q2) were recalculated. Note that the events of mutation from A to a or from a to A are independent in each generation in the simulation. Under neutral models (assuming only drift and mutation), we assumed that genetic variation at locus A in the total population was able to reach a steady polymorphic state, rather than being lost or fixed. Thus the same order of drift and mutation effects was employed in parameter settings. When the effects of selection were included in the model, gene frequencies were calculated according to the conventional method in each subpopulation. Finally, random sampling was conducted in both subpopulations (Nep and Ne(1−p) individuals for the MT-1 and MT-2 subpopulations, respectively). Population genetic differentiation (FstM) and LD were then calculated at each generation. Programs in C from Press et al. (1991) were employed for generating random numbers with a uniform distribution in the range (0, 1) and with a Poisson distribution. One thousand independent datasets were generated, and each was used to estimate FstM and LD. Means and standard deviations of estimated parameters were calculated from these replicated datasets.
Effects of the frequency of sexual reproduction
To evaluate the effects of different frequencies of sexual reproduction on FstM, we fixed all parameters (e.g., Ne = 400, Neμ = 1.0, Nev = 0.8, r = 0.5, p = 1/2) except the frequency of sexual reproduction s, which was varied across the range between 0 and 1.0. Figure 2 shows how at equilibrium genetic differentiation between subpopulations gradually decreases as the frequency of sexual reproduction increases. Theoretical predictions of FstM are in good agreement with the simulation results (Fig. 2a). As expected, the average LD is not different from zero, i.e. E(D) = 0.0, but the standard deviation decreases as the frequency of sexual reproduction increases (Fig. 2b). These simulation results imply that appropriate estimates of s can be derived from measurement of FstM if the effective population size (Ne) is known.

Estimates of FstM under different frequencies of sexual reproduction (s). a Mean and standard deviation of simulated FstM at steady state; b standard deviation for linkage disequilibrium (LD) at steady state. The steady-state results at each point are derived from 1000 independent simulations each for 1300 generations. Parameter settings are the total population size Ne = 400, proportion of the population with mating MT-1 type p = 0.5, the recombination rate r = 0.5 and the scaled mutation rates Neµ = 1.0 and Nev = 0.8
Effects of the effective population size
To evaluate the effects of effective population size on estimation of FstM, we fixed all parameters (e.g. p = 1/2, μ = 2.5 × 10−3, v = 2 × 10−3, r = 0.5, s = 0.05) while allowing Ne to vary between 50 and 2000. Under the neutral process, after equilibrium is reached, Fig. 3a shows how genetic differentiation between subpopulations gradually decreases as the effective population size increases. Theoretical predictions of FstM are in good agreement with the simulation results. When the genetic drift effect is much greater than mutation rates (1/Ne » μ or v), the mean FstM from simulations is smaller than the predicted result. However, all simulations are within the range of one standard deviation from theoretical predictions (Fig. 3a). The standard deviation of LD generally decreases as the effective population size increases (Fig. 3b). In general, simulation results indicate that appropriate estimates of FstM can be derived under weak or strong genetic drift effects.

Effects of effective population size (Ne) on estimation of FstM. a Mean and standard deviation of simulated FstM at steady state; b standard deviation for linkage disequilibrium (LD) at steady state. The steady-state results at each point are derived from 1000 independent simulations each for 1300 generations. Parameter settings are the proportion of the population with mating MT-1 type p = 0.5, the recombination rate r = 0.5, the probability of sexual reproduction s = 0.05 and the mutation rates µ = 2.5 × 10−3 and v = 2.0 × 10−3
Effects of recombination rate under neutrality
Figure S1 (Supplementary Materials) shows the approach to steady state of FstM and LD together with their standard deviations for different values of r. Figure 4a shows good agreement at steady state between the simulated and expected values of FstM over the full range of recombination rates. The predicted FstM vs. simulation results at steady state are 0.1163 vs. 0.0915 ± 0.1076 for r = 0.05, 0.0407 vs. 0.0421 ± 0.0544 for r = 0.25, and 0.0224 vs. 0.0246 ± 0.031 for r = 0.5, within one standard deviation of the simulation results. The steady-state LD is equal to zero. The simulated standard deviations for both FstM and LD increase as the A locus becomes more closely linked to the MT locus (Fig. 4a, b).

Effects of recombination rate (r) on estimation of FstM. a. Mean and standard deviation of simulated FstM at steady state; b. standard deviation for linkage disequilibrium (LD) at steady state. Results are derived from 1000 independent simulations with parameters of the total population size Ne = 400, proportion of the population with mating MT-1 type p = 0.5, probability of sexual reproduction s = 0.05 and scaled mutation rate Neµ = 1.0 and Nev = 0.8
Effects of mating-type frequencies
Figure S2 shows the approach to steady state when there are various degrees of asymmetry in mating-type subpopulation size (p ≠ 1/2). Figure 5a shows reduction in both the expected and simulated values of FstM as deviations from p = 1/2 occur. The theoretical predictions of FstM are consistent with the average estimates in the simulation results and are within the range of one standard deviation. The asymmetry does not affect the average LD, which is equal to the theoretical prediction, i.e. E(LD) = 0.0, but does reduce its standard deviation (Fig. 5b).

Effects of the frequency of mating types (p) on genetic differentiation between mating-type subpopulations FstM. a Mean and standard deviation of simulated FstM at steady state; b standard deviation for linkage disequilibrium (LD) at steady state. Results are derived from 1000 independent simulations, with parameter settings of the total population size Ne = 400, the recombination rate between A and mating-type loci r = 0.5, probability of sexual reproduction s = 0.05 and scaled mutation rate Neµ = 1.0 and Nev = 0.8
Effects of mutation rate
To simulate the effects of mutation rate on estimation of FstM, we fixed all parameters (p, Ne, r and s) except the mutation rate. For simplicity, we let the mutation rate from A to a and from a to A be equal, i.e. µ = v. Also, we considered symmetry between mating-type subpopulations (p = 1/2), unlinked loci (r = 0.5), the effective population size Ne = 1000 and the frequency of sexual reproduction s = 0.05. These parameter settings are arbitrary but biologically meaningful. The minimum mutation rate is set at the same order as the drift effects so that an equilibrium can be attained between drift and mutation. Figure 6a shows that mean FstM increases as mutation rate decreases. Theoretical predictions of FstM are greater than the simulation results although they are within the range of one standard deviation (Fig. 6a). The standard deviation of LD decreases as the mutation rate increases (Fig. 6b).

Effects of mutation rate on estimation of FstM. a Mean and standard deviation of simulated FstM at steady state; b standard deviation for linkage disequilibrium (LD) at steady state. The steady-state results at each point are derived from 1000 independent simulations each for 1300 generations. Parameter settings are the proportion of the population with mating MT-1 type p = 0.5, the recombination rate r = 0.5, the probability of sexual reproduction s = 0.05, and the effective population size Ne = 1000
Effects of selection on locus A
In the presence of deterministic selection, we consider a general case. Let the fitness of the four haplotypes be 1−x1, and 1−x2, respectively, for A and a on the MT-1 chromosome and 1−x3 and 1−x4, respectively, for A and a on the MT-2 chromosome, where xi (i = 1, 2, 3, 4) is the selection coefficient. Thus, compared with the preceding simulations, an additional process (selection) is assumed to operate on both the total population and each of the two subpopulations.
The first set of simulations modelled directional selection where either allele A or a was selectively advantageous irrespective of the mating-type subpopulation in which it was present. Results indicated that strong directional selection leads to a reduction in population genetic differentiation FstM compared to the neutral case but no departure of average LD from zero (Fig. 7a, Table S1)).

Effects of various forms of selection (selection coefficients xi = 0.002, 0.02 and 0.05) on estimates of population genetic differentiation between mating-type subpopulations (FstM) at a locus A linked to the MT locus with recombination rates r = 0.05 (left) and r = 0.05 (right). Values are derived from 1000 independent simulations at their steady-state distributions. a Directional selection. b Disruptive selection. c Stochastic selection with probability of an allele being favoured set at 0.2 or 0.8 (open squares) or 0.5 (filled squares). Expectations under neutrality (xi = 0) are given for comparison
We next modelled the case of disruptive selection, where allele A is selectively advantageous in the MT-1 subpopulation and allele a is advantageous in the MT-2 subpopulation (and vice versa). Results indicated that population genetic differentiation may be larger than that under the neutral process, especially when selective effects are greater than drift effects (selection coefficients >1/Ne, Fig. 7b, Table S1). When alleles initially at high frequency on appropriate mating-type chromosomes are selectively favoured, the initial coupling linkage phase is maintained even though recombination via sexual reproduction reduces LD. When selectively favoured alleles are initially at low frequency on appropriate mating-type chromosomes, the initial coupling linkage phase is eventually altered to the repulsion linkage phase through recombination by sexual reproduction (Table S1). Values of LD significantly above zero are maintained by such disruptive selection.
The final set of simulations modelled stochastic selection at locus A. Here the parameter α was used to set the probability that allele A was selectively advantageous in any one generation (x1 and x3 are set to zero, but x2 and x4 have positive values), while the probability that a was favoured in any generation was (1−α) (x2 and x4 are set to zero, but x1 and x3 have positive values). When selection coefficients were of the same order as the drift effects, a low (α = 0.2) or high (α = 0.8) probability of stochastic selection against allele A did not significantly reduce population genetic differentiation, compared with the result under neutrality (Fig. 7c, Table S1). However, when selection was stronger (xi > 1/Ne), a significantly reduced level of population genetic differentiation (FstM) was found compared with that under neutrality. When there was an equal probability of selection against allele A and a per generation, population genetic differentiation was essentially the same as that under neutrality, irrespective of weak or strong selection (Fig. 7c, Table S1).
Inferring the number of sexual outcrosses per generation
Application to existing data
To investigate the utility of the new method for estimating the number of individuals per generation participating in sexual reproduction in facultatively sexual haploid populations, we analysed a number of existing datasets from ascomycete fungi in which population samples had been scored both for the mating-type locus and a set of molecular markers. These markers are assumed to be selectively neutral and unlinked to the mating-type locus. Datasets were from heterothallic ascomycetes in which the mating-type ideotypes were found at equal frequency. They comprised two samples collected early and late in the same season from a population of Zymoseptoria tritici and scored for restriction fragment length polymorphism markers (Chen and McDonald 1996); two samples of Erisiphe necator, collected late in one season and early in the next season from the same population and scored for microsatellite markers (Brewer et al. 2012); two populations of Rhyncosporium secalis scored for microsatellite markers (Linde et al. 2003; Linde et al. 2009); and one population of Dothistroma septosporum scored for microsatellite markers (Piotrowska et al. 2018).
For each population analysed, Χ2 tests with one degree of freedom were used to determine the significance of departures from a 1:1 ratio of mating types. The program MLGsim v.2.0 (Stenberg et al. 2003) was then used to recognise clonal replicates within the total dataset. Subsequent analyses were conducted on both the original data and on a clone corrected dataset.
To estimate FstM and its statistical significance, populations were divided into MT-1 and MT-2 subpopulations. Genetic differentiation between these subpopulations at the marker loci scored was determined using Weir and Cockerham’s (1984) estimator of Fst implemented in FSTAT v2.9.3.2 (Goudet 2002). Where significant values of FstM were found, these were used to estimate the number of sexual events per generation (Nes) using Eq. (6) and its standard error using Eq. (7). Given the sample size, the number of markers and genetic diversity of the datasets analysed, it is likely that the smallest significant value of FstM that can be detected is close to 0.01. Therefore, where estimates of Fst were statistically non-significant we inferred that the value of FstM lay below 0.01, and in this situation our estimated value of Nes was taken to be >50, according to Eq. (6).
To compare our analysis based on FstM with measures of LD that are commonly used to infer rates of sexual reproduction (e.g. Brewer et al. 2012), we also used FSTAT to calculate pairwise LD values between marker loci and their significance within each population after Bonferroni correction for multiple tests. Finally, the program MultiLocus v.1.3b (Agapow and Burt 2001) was employed to determine the correlation coefficient among loci based on gene frequencies rD, a standardised measure of genome-wide LD that is independent of the number of loci scored (Brown et al. 1980; Maynard Smith et al. 1993; Agapow and Burt 2001). The significance of rD was determined using 1000 random permutations of the data.
Results
Analyses of the number of sexual reproduction events per generation in the four ascomycete species are summarised in Table 1. For Z. tritici, both early and late samples showed no significant genetic differentiation between mating-type subpopulations both before and after clone correction, implying that large numbers of individuals (>50) are involved in sexual reproduction each generation. These conclusions are supported by the very low percentage of pairwise LD found in both populations. However, in the early sample highly statistically significant, though numerically low, values of rD were found.
For the sample of E. necator taken late in the season, there was significant differentiation between mating-type subpopulations both before (FstM = 0.201**, P value < 0.01) and after (FstM = 0.050*, P value < 0.05) clone correction, and the numbers of individuals involved in sexual reproduction each generation were estimated as 2.0 ± 0.6 and 9.5 ± 4.6, respectively. These values were accompanied by a high percentage of loci showing LD and large and significant values of rD. In contrast, for the same population of E. necator sampled early the next season, mating-type subpopulations showed no evidence for genetic differentiation either before or after clone correction, implying a large number of individuals (>50) involved in sexual reproduction each generation. However, over 10% of locus pairs showed significant LD and rD was significant and twice as high as in the Z. tritici samples.
The two samples of R. secalis displayed very similar patterns with significant genetic differentiation between mating-type subpopulations before (FstM = 0.043** for the Norway sample and 0.149** for the Australia sample) but not after clone correction. Estimates of the number of sexually reproducing individuals lay between 2.9 ± 0.5 (minimum estimate from original data) and >50 (maximum estimate from clone corrected data). Overall, this suggests a lower number of sexually reproducing individuals per generation than for either Z. tritici or E. necator. Clone correction reduced both the high percentage of locus pairs showing LD and the large values of rD found in the original samples, although these remained substantial.
Finally, in the single population of D. septosporum analysed, significant differentiation between mating-type subpopulations was found both before (FstM = 0.107**) and after (FstM = 0.022*) clone correction. Minimum and maximum estimates of the number of sexually reproducing individuals were 4.2 ± 1.1 and 22.2 ± 10.3, respectively, lower than for any of the species previously analysed. The percentage of locus pairs showing LD was substantial, and values of rD were high and significant both in the initial sample and after clone correction.
Discussion
In this paper, we have developed a simple genetic model for estimating Nes, the number of sexual reproduction events that occur each generation within facultatively sexual haploid populations possessing two mating types. The model is applicable to populations in which there are equal frequencies of the two mating types, a situation which already implies the presence of some sexual reproduction; the novelty of the model is that it allows quantification of Nes. The model requires data on the genotype of individuals both at the mating-type locus and at a number of selectively neutral markers that are unlinked to the mating-type locus. Application of the model to existing data from ascomycete populations suggests high values of Nes in Z. tritici and E. necator, intermediate levels in R. secalis and low levels in D. septosporum.
The model that we have developed has a number of limitations that need to be appreciated whenever it is applied. First, it assumes that genetic differentiation between mating-type subpopulations FstM is accounted for by a drift–migration–mutation equilibrium. Estimates of Nes will therefore be long-term average estimates rather than estimates for contemporary populations. This contrasts with alternative analyses based on frequency of clonal recapture that provide estimates of sexual reproduction frequency for contemporary populations (Ali et al. 2016). Also implicit in our model is that the mating type of an individual is fixed and individuals cannot transfer, by mating-type switching, from one mating type subpopulation to the other (Perkins 1987; Nieuwenhuis and Immler 2016).
Two further assumptions of the method are that the marker loci used to estimate FstM are neither linked to the mating-type locus nor subject to selection. We have shown that linkage to the mating-type locus enhances the expected value of FstM, while selection on the marker loci may either reduce (directional selection) or increase (disruptive selection) the expected value. In practise, when applying the analysis to non-model organisms, it may be difficult to test these assumptions. A step that could be used to filter out inappropriate marker loci from an analysis would be to compare FstM values among loci and remove those that generate outliers.
A final assumption of our model is that each new generation is founded from a mixture of simultaneously generated sexually and asexually reproduced individuals (Fig. 1). This is appropriate for the ascomycete species used to test the validity of our approach. However, our model is unsuitable for analysing situations where a series of purely asexual generations are interspersed by one or more generations of synchronous sexual reproduction. Here alternative models would have to be developed to estimate the average proportion of all generations that were sexual.
Our estimates of Nes in the four ascomycete species analysed are consistent with the known biology of the taxa concerned. Sexual ascospores are believed to be the primary form of inoculum for each new generation of both Z. tritici and E. necator in the regions from which our samples were derived (Suffert et al. 2011; Pearson and Gadoury 1987). Our analyses suggest high values of Nes for both taxa, at least in early season samples. In R. secalis, the presence of sexual reproduction has been inferred from previous studies of genetic structure and mating-type frequencies, but sexual fruiting bodies have not been identified in the field (McDonald et al. 1999; Salamati et al. 2000; Linde et al. 2003). This is consistent with the lower estimates of Nes for the species in our analysis. Finally, in British populations of D. septosporum, the primary source of inoculum in each generation is known to be asexual conidia (Mullett et al. 2016), although the sexual fruiting body has occasionally been found in continental Europe (Butin 1985). A low value of Nes, as inferred from our analysis, is therefore to be anticipated.
A result which is inconsistent with our expectations is that for the late season sample of E. necator. Here a significant value of FstM was detected. Such differentiation was absent in a sample from the same populations taken early in the next season, which is thought to have been founded entirely from sexual offspring (Brewer et al. 2012). One explanation for the late season result may be that after establishment the population has been subject to strong selection favouring particular clones (selection coefficient » 1/Ne) leading, as a consequence of clonal hitchhiking, to large differences in marker allele frequency between the mating-type subpopulations. This is consistent with previous detection of large, spatially structured clones in this population (Brewer et al. 2012) and the present clone correction analysis that found only 48 distinct clones among a sample of 78 isolates (Table 1). This implies that, in order to obtain reasonable estimates of Nes in situations where strong selection may be operating, it will be important to obtain samples from early in each generation before allele frequencies in mating-type subpopulations have been affected by unequal clonal expansion. If samples are not available from early in the generation, a clone correction could be applied to the data collected later in the season (Arnaud-Haond et al. 2007). The objective would be to collapse genotypes isolated multiple times, and assumed to be the product of asexual reproduction during the season, into a single genotype. However, if the founding population had originally contained multiple asexually produced individuals of the same genotype, these too would be collapsed into a single genotype and lost from the analysis, inadvertently enhancing the estimate of Nes. Therefore, in the presence of strong selection it may be best to regard estimates of Nes from the original dataset as minimum estimates and those from the clone corrected dataset as maximum estimates.
The analysis described here estimates the absolute number of sexual events occurring in each generation in a facultatively sexual haploid population but does not allow the evolutionarily more important parameter, the frequency of sexual reproduction s, to be calculated. This requires an additional estimation of Ne, the effective size of the population. Ne could be calculated by sampling the target population twice, a known number of generations apart, and measuring the variance in marker allele frequencies between the two samples (Waples 1989). An additional advantage of adopting this strategy is that it would provide the opportunity independently to estimate s and Ne using the clone recapture technique of Ali et al. (2016) to allow a comparison with results from the present analysis.
In the examples that we have used to illustrate the application of our new method, genetic data were derived from a relatively limited number of loci, which we have assumed are unlinked to the mating-type locus. Consistency of FstM estimates over loci within each analysis suggest that the latter assumption is not a serious problem. However, the low number of loci scored means that there is limited power to detect significant genetic differentiation between mating-type subpopulations, placing upper limits on our ability to estimate Nes. In the future, there will be the opportunity to overcome these problems by making use of data from population samples of re-sequenced genomes (Grunwald et al. 2016; Moller and Stuckenbrock 2017). Such analyses yield very large numbers of SNP markers with known linkage relationships to the mating-type locus and should allow far more precise estimates of Nes using the model developed here.
Data archiving
Population genetic data used to analyse sexual reproduction and the code used for simulations have been submitted to Dryad (https://doi.org/10.5061/dryad.3p4v855).
References
Agapow PM, Burt A (2001) Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1:101–102
Ali S, Gladieux P, Rahman H et al. (2014) Inferring the contribution of sexual reproduction, migration and off-season survival to the temporal maintenance of microbial populations: a case study on the wheat fungal pathogen Puccinia striiformis f.sp. tritici. Mol Ecol 23:603–617
Ali S, Soubeyrand S, Gladieux P, Giraud T, LeConte M, Gautier A, Mboup M, Chen W, De Vallavieille-Pope C, Enjalbert J et al. (2016) CLONCASE: estimation of sex frequency and effective population size by clonemate resampling in partially clonal organisms. Mol Ecol Resour 16:845–861
Arnaud-Haond S, Duarte CM, Alberto F, Serrão EA (2007) Standardizing methods to address clonality in population studies. Mol Ecol 16:5115–5139
Billiard S, Lopez-Villavicencio M, Hood ME, Giraud T (2012) Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J Evol Biol 25:1020–1038
Brewer MT, Frenkel O, Milgroom MG (2012) Linkage disequilibrium and spatial aggregation of genotypes in sexually reproducing populations of Erysiphe necator. Phytopathology 102:997–1005
Brown ADH, Feldman MW, Nevo E (1980) Multilocus structure of natural populations of Hordeum spontaneum. Genetics 96:523–536
Butin H (1985) Development of the teleomorph and anamorph of Scirrhia pini Funk & Parker on needles of Pinus nigra Arnold. Sydowia 38:20–27
Chen RS, McDonald BA (1996) Sexual reproduction plays a major role in the genetic structure of populations of the fungus Mycosphaerella graminicola. Genetics 142:1119–1127
Constable GWA, Kokko H (2018) The rate of facultative sex governs the number of expected mating types in isogamous species. Nat Ecol Evol 2:1168–75
Goddard MR, Godfray HCJ, Burt A (2005) Sex increases the efficacy of natural selection in experimental yeast populations. Nature 434:636–640
Goudet J (2002). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). Updated from Goudet (1995). Available from: <http://www2.unil.ch/popgen/softwares/fstat.htm> (accessed Jul 2018).
Grunwald NJ, McDonald BA, Milgroom MG (2016) Population genomics of fungal and oomycete pathogens. Ann Rev Phytopath 54:323–346
Hadjivasiliou Z, Pomiankowski A (2016) Gamete signalling underlies the evolution of mating types and their number. Philos Trans R Soc B 371:20150531
Hartfield M, Wright SI, Agrawal AF (2018). Coalescence and linkage disequilibrium in facultatively sexual diploids. Genetics 210:683–701.
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231
Lehtonen J, Jennions MD, Kokko H (2012) The many costs of sex. Trends Ecol Evol 27:172–178
Linde CC, Zala M, Ceccarelli S, McDonald BA (2003) Further evidence for sexual reproduction in Rhynchosporium secalis based on distribution and frequency of mating-type alleles. Fungal Genet Biol 40:115–125
Linde CC, Zala M, McDonald BA (2009) Molecular evidence for recent founder populations and human-mediated migration in the barley scald pathogen Rhynchosporium secalis. Mol Phylogenet Evol 51:454–464
Lopez-Villavicencio M, Schoustra S, Giraud T, Debets AJM (2013) Evidence for deleterious effects of recombination and advantages of sex independent of recombination using fungal models. J Evol Biol 26:1968–1978
May G, Shaw F, Badrane H, Vekemans X (1999) The signature of balancing selection: fungal mating compatibility gene evolution. Proc Natl Acad Sci USA 96:9172–9177
Maynard Smith J, Smith NH, O’Rourke M, Spratt BG (1993) How clonal are bacteria? Proc Natl Acad Sci USA 90:4384–4388
McDonald BA, Zhan J, Burdon JJ (1999) Genetic structure of Rhynchosporium secalis in Australia. Phytopath 89:639–645
de Meeus T, Prugnolle F, Agnew P (2007) Asexual reproduction: genetics and evolutionary aspects. Cell Mol Life Sci 64:1355–1372
Milgroom MG (1996) Recombination and the multilocus structure of fungal populations. Ann Rev Phytopathol 34:457–477
Moller M, Stuckenbrock EH (2017) Evolution and genome architecture in fungal plant pathogens. Nat Rev Microbiol 15:756–771
Muller HJ (1964) The relation of recombination to mutational advance. Mutat Res 106:2–9
Mullett MS, Tubby KV, Webber JF, Brown AV (2016) A reconsideration of natural dispersal distances of the pine pathogen Dothistroma septosporum. Plant Pathol 65:1462–1472
Nieuwenhuis BPS, James TY (2016) The frequency of sex in fungi. Philos Trans R Soc B 371:20150540
Nieuwenhuis BPS, Immler S (2016) The evolution of mating-type switching for reproductive assurance. BioEssays 38:1141–1149
Ohta T, Kimura M (1969) Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics 63:229–238
Ohta T, Kimura M (1970) Development of associative overdominance through linkage disequilibrium in finite populations. Genet Res 16:165–177
Pearson RC, Gadoury DM (1987) Cleistothecia the source of primary inoculum for grape powdery mildew in New York USA. Phytopathology 77:1509–1514
Perkins DD (1987) Mating-type switching in filamentous ascomycetes. Genetics 115:215–216
Piotrowska MJ, Riddell C, Hoebe PN, Ennos RA (2018) Planting exotic relatives has increased the threat posed by Dothistroma septosporum to the Caledonian pine populations of Scotland. Evol Appl 11:350–363
Press WH, Flannery BP, Teukolsky SA, Vetterling WT (1991) Numerical recipes in C: the art of scientific computing. Cambridge University Press, Cambridge
Salamati S, Zhan J, Burdon JJ, McDonald BA (2000) The genetic structure of field populations of Rhynchosporium secalis from three continents suggests moderate gene flow and regular recombination. Phytopathology 90:901–908
Siah A, Tisserant B, El Chartouni L, Duyme F, DeWeer C, Roisin-Fichter C, Sanssene J, Durand R, Reignault P, Halama P (2010) Mating type idiomorphs from a French population of the wheat pathogen Mycosphaerella graminicola: widespread equal distribution and low but distinct levels of molecular polymorphism. Fungal Biol 114:980–990
Stenberg P, Lundmark M, Saura A (2003) MLGsim: a program for detecting clones using a simulation approach. Mol Ecol Notes 3:329–331
Suffert F, Sache I, Lannou C (2011) Early stages of Septoria tritici blotch epidemics of winter wheat: build-up, overseasoning, and release of primary inoculum. Plant Pathol 60:166–77
Talas F, McDonald BM (2015) Genome-wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genom 16:966
Taylor JW, Hann-Soden C, Branco S, Sylvain I, Ellison CE (2015) Clonal reproduction in fungi. Proc Natl Acad Sci USA 112:8901–8908
Tsai IJ, Bensasson D, Burt A, Koufopanou V (2008) Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. Proc Natl Acad Sci USA 105:4957–4962
Waples RS (1989) A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121:379–391
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Wright S (1969) Evolution and the genetics of populations. Vol. 2: The theory of gene frequencies. The University of Chicago Press, Chicago
Acknowledgements
We are very grateful to Bruce McDonald and to Marin Talbot Brewer and Michael Milgroom for access to their population genetic data on Z. tritici and R. secalis and on E. necator, respectively, without which this paper would not have been possible. Population genetic data on D. septosporum came from the PROTREE project funded jointly by a grant from BBSRC, Defra, ESRC, the Forestry Commission, NERC and the Scottish Government, under the Tree Health and Plant Biosecurity Initiative. XSH is supported by funding from the South China Agricultural University (4400-K16013). We thank the editor and three anonymous referees for constructive comments that greatly improved the original manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Ennos, R.A., Hu, XS. Estimating the number of sexual events per generation in a facultatively sexual haploid population. Heredity 122, 729–741 (2019). https://doi.org/10.1038/s41437-018-0171-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0171-1
