
Abstract
Epigenetics refers to chemical modifications of chromatin or transcribed DNA that can influence gene activity and expression without changes in DNA sequence. The last 20 years have yielded breakthroughs in our understanding of epigenetic processes that impact many fields of biology. In this review, we discuss how epigenetics relates to quantitative genetics and evolution. We argue that epigenetics is important for quantitative genetics because: (1) quantitative genetics is increasingly being combined with genomics, and therefore we should expand our thinking to include cellular-level mechanisms that can account for phenotypic variance and heritability besides just those that are hard-coded in the DNA sequence; and (2) epigenetic mechanisms change how phenotypic variance is partitioned, and can thereby change the heritability of traits and how those traits are inherited. To explicate these points, we show that epigenetics can influence all aspects of the phenotypic variance formula: VP (total phenotypic variance) = VG (genetic variance) + VE (environmental variance) + VGxE (genotype-by-environment interaction) + 2COVGE (the genotype–environment covariance) + Vɛ (residual variance), requiring new strategies to account for different potential sources of epigenetic effects on phenotypic variance. We also demonstrate how each of the components of phenotypic variance not only can be influenced by epigenetics, but can also have evolutionary consequences. We argue that no sources of epigenetic effects on phenotypic variance can be easily cast aside in a quantitative genetic research program that seeks to understand evolutionary processes.
Similar content being viewed by others


Impacts of epigenetics research
Epigenetics refers to chemical modifications of chromatin or transcribed DNA that can influence gene activity and expression without changes in DNA sequence (Jablonka and Raz 2009; Kilvitis et al. 2014). The most well-studied epigenetic mechanism is the methylation of cytosines within DNA sequences (Rapp and Wendel 2005; Allis and Jenuwein 2016; Richards et al. 2017), although other mechanisms like histone modifications (Berger et al. 2009) and a heterogeneous assortment of RNA regulatory systems (Chen et al. 2011) have also been uncovered and studied intensively (reviewed in Allis and Jenuwein 2016). Methylation of cytosines is an important component of silencing transposable elements (Gibney and Nolan 2010; Huff and Zilberman 2014; Fultz et al. 2015; Ikeda and Nishimura 2015), but is also found in complex patterns across other genomic contexts (Niederhuth et al. 2016). For example, dense gene promoter methylation is associated with silencing of genes, but methylation in gene bodies—which tends to be polymorphic between individuals—is poorly associated with gene expression and varies widely across taxa (Becker et al. 2011; Nätt et al. 2012; Schmitz et al. 2013; Niederhuth and Schmitz 2017).
Epigenetics research has surged over the last 20 years, thanks to advances in our knowledge about chromatin modifications and their modifiers, and the often-complex connections between these modifications and gene expression patterns (reviewed in Allis and Jenuwein 2016). In particular, “the modern era of epigenetic research” is transforming our understanding of the molecular basis of development and disease (Allis and Jenuwein 2016). But the integration of epigenetics into evolutionary thinking is still gathering momentum (Laland et al. 2015; Burggren 2016; Richards et al. 2017). Epigenetics is one of several new research topics that are emerging in evolutionary biology that go beyond the original tenets of the Modern Synthesis (Huxley 1942), the major integration of disparate biological and mathematical concepts into evolutionary theory in the mid-20th century (Pigliucci 2007; Plutynski 2009; Pigliucci and Müller 2010; Robertson and Richards 2015).
Proponents of a so-called “Extended Evolutionary Synthesis” are dissatisfied with a definition of evolution that is limited to changing DNA sequence based on allele frequencies over time. Instead, the evolutionary process would be more completely described by investigating non-genetic mechanisms like epigenetics that were not initially included in the Modern Synthesis, and that might be able to overcome some of the limitations of strictly DNA sequence-based inheritance (Pigliucci 2007; Bonduriansky and Day 2009; Day and Bonduriansky 2011; Robertson and Richards 2015). In particular, because epigenetic mechanisms are more dynamic and reversible than DNA sequence, epimutations have been implicated as a faster source of adaptation than genetic mutations (Jablonka and Lamb 1989, Jablonka et al. 1998). Epimutations can provide higher phenotypic variance at equilibrium due to different epigenetic states, and enhance the adaptive possibilities of asexual or low-diversity taxa (Jablonka and Lamb 1989; Geoghegan and Spencer 2012, 2013). Organisms may use the additional variation afforded by epigenetic mechanisms as a bet-hedging strategy in unknown environments (Jablonka and Lamb 1989, Jablonka et al. 1998; Pál and Miklós 1999). Epigenetic modifications could also "hold" a potentially advantageous phenotype for multiple generations, allowing time for more stable genetic variants to stabilize the phenotype (i.e. canalization or genetic assimilation, Waddington 1942, 1953; West-Eberhard 2005; Klironomos et al. 2013; Kronholm and Collins 2016).
Epigenetics should be incorporated into evolutionary theory, because the molecular underpinnings of phenotypic variation are the subject of many evolutionary research programs (McNiven et al. 2011). We simply need to expand our thinking to include epigenetic mechanisms, which are another class of molecular mechanisms. Evolution relies on heritable phenotypic variation, which is provided by alleles (which may be DNA-based alleles or epigenetic-based alleles, as we will describe) that confer different patterns of gene expression/function that ultimately result in different phenotypes in different individuals (Falconer and Mackay 1996). Roff (2007) and Hill (2012) took stock of evolutionary biology in the genomics era, where the advent of high-throughput genome sequencing technologies of the last few decades (e.g., Liberles 2001; Goodwin et al. 2016) has been revolutionary across the life sciences. They point out that even quantitative genetics, a branch of evolutionary biology that statistically analyzes phenotypes and has treated molecular mechanisms as a black box, has been greatly influenced by the genomic era, with many research programs now focusing on the number and distribution of effect sizes of loci affecting a trait in a population through the use of quantitative trait locus (QTL) or genome-wide association mapping (GWAM) (Li et al. 2016). Genomics approaches have also provided new resolution to understanding the interactions among loci, and the change in the phenotypic effects of loci under different environments (Mackay 2001). High-throughput genome sequencing technologies have enabled the evolutionary biology community to blend quantitative genetics and genomic approaches to address the enduring question about whether phenotypic variation is controlled by many loci of small effect, fewer loci with a wider distributions of effect sizes, or just a few loci of large effect on the phenotype (Roff 2007; Kalisz and Kramer 2008; Stinchcombe and Hoekstra 2008; Hill 2012). Further, we now have more power to ask whether loci are behaving in an additive or more complex fashion in their phenotypic effects (Ruppell et al. 2004; Wolf et al. 2005; Mackay 2014).
The knowledge of the genetic architecture of traits can be used to study many aspects of evolution. For instance, genetic architecture can provide evidence for the degree to which negative selection, stabilizing selection, and balancing selection shaped the evolution of a trait, as reflected in the distributions of the minor allele frequencies of the QTLs/SNPs, as well as in the distribution of effect sizes of the QTLs/SNPs (Erickson et al. 2004; Josephs et al. 2017). Knowledge of the genetic architecture can also be leveraged in artificial selection. Agricultural breeding programs currently make prodigious use of a technique known as genomic selection to improve plant and animal breeding (Meuwissen et al. 2001; Koopaee and Koshkoiyeh 2014). The approach first uses GWAM to find associations between molecular markers and the phenotype(s) of interest in crops or livestock. Then, individuals with unknown phenotype or pedigree are genotyped at the same SNP sites (using a low-cost SNP chip) to forecast their phenotype(s) from the effect sizes associated with each allele from the GWAM study (Koopaee and Koshkoiyeh 2014). In this way, superior stock can be selected for breeding and development without further measuring of phenotype. In addition to this type of application, genomic selection has been used in model systems to make more precise predictions about adaptive evolution in response to selection (e.g., Edwards et al. 2016; Kooke et al. 2016).
Epigenetics and the missing heritability problem
A well-known problem with genetic mapping is that not all of the genetic variance (VG) in the phenotype can be accounted for by variance in the genome, a phenomenon known as the “missing heritability” problem (Brachi et al. 2011; Caballero et al. 2015). In QTL studies of recombinant inbred lines (RIL) or GWAM studies of natural variants, some differences in phenotypes cannot be accounted for by the sum of the significantly correlated genomic regions. Multiple explanations have been put forth to account for the missing heritability (Dickson et al. 2010; Rockman 2012; Hemani et al. 2013; Yang et al. 2015). Epigenetic changes are another possible source of some of the missing heritability, because epigenetic changes can be inherited and act in an additive fashion on phenotypic variance (Morgan et al. 1999; Henderson and Jacobsen 2007; Slatkin 2009; Bell and Spector 2011; González-Recio 2011; Migicovsky and Kovalchuk 2011; Goddard and Whitelaw 2014; Triantaphyllopoulos et al. 2016). Epigenetic changes that have phenotypic effects (mediated through their effects on gene expression) and that are also inherited could generate associations between individuals and phenotypes that may not be explained based on variation in DNA sequence-based markers.
There are many examples of epigenetically inherited effects that could account for missing heritability. One particularly illustrative example comes from the agouti locus in mice (Morgan et al. 1999; Waterland and Jirtle 2003). Differences in silencing by methylation of a retrotransposon inserted upstream of the Avy allele at the agouti locus is correlated with ectopic agouti expression and disease risk. Low methylation of the transposable element is correlated with yellow coat color and diseases such as obesity, diabetes, and tumors, while high methylation of the transposable element is associated with brown coat color and lessened disease risk (Morgan et al. 1999; Waterland and Jirtle 2003). Importantly, specific agouti epialleles in female mice can be induced by a high methyl donor diet, and the epigenetic state of the agouti allele is incompletely reset by meiosis in the female germ line (Morgan et al. 1999; Waterland and Jirtle 2003). Thus, coat color, along with susceptibility to cancer and metabolic disorders, can be induced in the mother and inherited by the offspring, illustrating that epigenetic changes can be inherited in a manner like DNA sequence-based mutations, but can also be induced in a specific, directional way by the environment.
Several other studies in ecological epigenetics provide examples of epigenetic effects that are inherited (Sano and Kim 2013; Robertson and Richards 2015; Richards et al. 2017). Verhoeven et al. (2010) reared apomictic dandelions (Taraxacum officinale) under several different stresses (nutrient, salt, and chemical stresses). They found that the stresses triggered considerable methylation variation throughout the genome, and many of these were faithfully transmitted to the offspring. These results demonstrated that epigenetic variation can be readily generated by environmental challenges and that those changes can be inherited. In another example, Richards et al. (2008, 2012) demonstrated the persistence of epigenetic differences in plants from the Fallopia species complex (referred to as Japanese knotweed sensu lato) with little DNA sequence variation. The Fallopia species complex occupies a wide range of habitats in Europe and has colonized an even wider array of habitats in the northeastern US (including marshes and beaches). Richards et al. (2012) found that the invasive Fallopia at sites on Long Island in New York, USA had only four variable AFLP positions out of 200 positions genome-wide. They found diversity was five times higher at epigenetic loci, and this diversity varied among sites. Importantly, these data were collected from leaf tissue that was grown from rhizomes in a common garden. Therefore, the DNA methylation patterns were not merely induced by different sites, but persisted in tissue that was created in a common environment. Perhaps not all of this epigenetic variation would be inherited through meiosis, but some of it could be as has been found in other studies (Cubas et al. 1999; Feng et al. 2010; Herrera et al. 2013). More importantly, for the biology of this species, clonal propagation is a common mode of expansion and therefore the persistence of differences in DNA methylation through clonal propagation is highly relevant (Verhoeven and Preite 2014; Douhovnikoff and Dodd 2015; Rendina González et al. 2016; Spens and Douhovnikoff 2016).
Neither the Verhoeven et al. (2010) study nor the Richards et al. (2012) study correlated inherited epigenetic variation with phenotypic variation, but such linkages have been made in other studies (reviewed in Richards et al. 2017). Cubas et al. (1999), for instance, characterized a naturally occurring mutant of toadflax (Linaria vulgaris) that has a different floral symmetry pattern. They found that methylation changes, rather than DNA sequence changes, to a floral symmetry gene explained the phenotypic change. The changes in methylation of the gene segregate and are inherited much like DNA sequence variants, but spontaneous resetting of methylation sometimes occurs in nature that recovers the non-mutant phenotype.
In another example linking epigenetic variation and phenotypic variation, Johannes et al. (2009) developed a recombinant inbred line (RIL) population of the model plant Arabidopsis thaliana that segregates for differentially methylated positions (DMPs)—differences in whether individual cytosines are methylated or not. The epigenetic recombinant inbred lines (epiRILs) are nearly isogenic in terms of the DNA sequences, but show variation and high heritability for many traits relating to growth and morphology, including plant height, flowering time, and primary root length (Zhang et al. 2013; Cortijo et al. 2014; Kooke and Keurentjes 2015). The epiRILs were derived from crosses between the Columbia wild-type genotype and a mutant line derived from the Columbia wild-type with a mutation in the DECREASED DNA METHYLATION 1 (DDM1) locus. Mutant plants (ddm1/ddm1) exhibit 70% less methylation genome-wide than wild-type plants (Vongs et al. 1993). From this initial cross, Johannes et al. (2009) selected backcross progeny that were homozygous for the wild-type (DDM1/DDM1) at the DDM1 locus to propagate 505 epiRILs through six rounds of single-seed descent. Thus, the epiRILs are nearly isogenic in terms of DNA sequences, but segregate for stably inherited methylation polymorphisms (DMPs), which are homozygous within each epiRIL (Johannes et al. 2009). The epiRILs show phenotypic variation and high heritability for plant height, flowering time, and primary root length (Johannes et al. 2009; Cortijo et al. 2014; Kooke and Keurentjes 2015). Of particular interest, Cortijo et al. (2014) found multiple epiQTLs accounting for 60–90% of the heritability of flowering time and primary root length. Thirty percent of the heritable DMPs identified in the epiRIL population also overlapped with naturally occurring epipolymorphic regions among 138 natural A. thaliana accessions, suggesting that epigenetic variation could be important in the wild. These epiQTLs have all the necessary properties (stability, inheritance, variance, phenotypic effects) to be targets of natural or artificial selection.
Differences between epigenetic effects and parental effects
Parental effects refer to traits expressed in parents that influence traits expressed in offspring (Hadfield 2012). Under this umbrella are both paternal effects and maternal effects, allowing for fathers’ and mothers’ phenotypes to have different effects on offsprings’ phenotypes. Maternal effects have generally received the most theoretical attention, presumably because prenatal maternal provisioning and postnatal maternal care are the most significant sources of parental effects (Kirkpatrick and Lande 1989; Wolf and Wade 2016). Epigenetic and parental effects differ in that epigenetic effects are a more specific class of phenomena than parental effects; epigenetic effects result from specific types of cellular mechanisms, whereas parental effects can include any mechanism by which a trait expressed in the parents is also expressed in the offspring. Parental effects can be due to inherited epigenetic effects, but they can also be due to non-epigenetic mechanisms, like cultural transmission, prenatal nutrient provisioning, postnatal care, and maternal transmission of mitochondria, chloroplasts, and other cytoplasmic factors to the offspring (Kirkpatrick and Lande 1989; Wolf and Wade 2016). Epigenetics also differs from parental effects because epigenetic effects are not necessarily inherited, whereas parental effects are, by definition, inherited. Consider, for instance, a change in the methylation state at a particular region of a chromosome of an organism in response to some environmental stimulus, where the methylation changes are reset in the germ line. The methylation changes in this example would be epigenetic, but they would not be a parental effect. This is because, in this example, the methylation changes in the parents would not be transmitted to the offspring, and therefore the methylation changes in the parents would not serve as the vehicle through which “information” about the parental traits are passed to the offspring. In summary, parental effects and epigenetics are overlapping but also distinct concepts that should be modeled separately and can have different effects on the phenotype and on inheritance (Santure and Spencer 2006).
Differences between epigenetic effects and genetic effects
Epigenetic mechanisms can contribute to phenotypic variation and could be playing a role in the missing heritability of quantitative traits. But epigenetic contributions to heritability are different from DNA sequence based contributions to heritability, because epigenetic states can be reset, either before reaching the germ line or after transmission to the offspring (Saze et al. 2003; Feng et al. 2010; Hackett et al. 2013), and because epigenetic differences among individuals can arise at different rates from DNA sequence-based differences (Kronholm and Collins 2016). Modeling efforts have demonstrated that epigenetic effects have different ramifications for evolution than standard DNA sequence based processes. Furrow (2014), for instance, used a population genetic model to understand the response of epigenetically induced phenotypic variation to selection over multiple generations. He found that, because epimutation rates are high, the response to selection decays more rapidly across generations than would be expected based on the observed heritability of a trait alone. Similarly, Santure and Spencer (2006) found a smaller than expected response to selection when they included epigenetic differences that depended on the parent from whom a chromosomal region was inherited (genomic imprinting; Macdonald 2012). Thus, traits may be less responsive to natural selection than expected due to the effect of epigenetic phenomena on observed heritabilities (but see Klironomos et al. 2013). In addition to the rate of epimutation, Kronholm and Collins (2016) found that the effect size of the epimutations influences the rate of adaptation, where small-effect epimutations generally speed up adaptation, and large-effect epimutations slow down adaptation. In contrast to models that found that epigenetic effects may slow down adaptation, a model by Uller et al. (2015) showed that epigenetic effects can speed up evolution in heterogeneous but predictable environments, due to the environmental induction of some epigenetic states and their transmissibility. Another model by Leimar and McNamara (2015) showed that incompletely-resetting epigenetic systems are ideally suited for actively transmitting environmental information from previous generations, and can evolve readily. Thus, selection may be actively maintaining that the cellular machinery does not completely reset certain environmentally induced epigenetic states across generations, something that Shea et al. (2011) refer to as a “selection-based effect” of epigenetics.
Several authors have emphasized how the decoupling of phenotypic change from genotypic change enabled by epigenetic mechanisms can allow populations to change in ways that might not otherwise be possible. For instance, epigenetic changes that happen at faster rates than DNA sequence changes can drive more rapid evolutionary change by serving as a bridge to allow phenotypic changes to occur first, and genetic changes later (Bonduriansky and Day 2009; Geoghehan and Spencer 2013; Klironomos et al. 2013; Kronholm and Collins 2016). In addition, epimutations may allow for a higher mutational load and lower mean fitness of adapted populations than would otherwise be expected (due to the higher epimutation rate; Klironomos et al. 2013), but recent studies found that epigenetic variation may compensate for the negative effects of inbreeding (Vergeer et al. 2012; Liebl et al. 2013). When van der Graaf et al. (2015) combined theoretical modeling with high-resolution epigenetic analysis of multiple independent A. thaliana mutation accumulation lines, they found that the mean fitness due to epimutations was not lower than expected. The epimutation rates they reported were high enough to rapidly uncouple genetic from epigenetic variation, but low enough for new epialleles to sustain long-term selection responses. We will need more information before we can make conclusions about the specific ramifications of epigenetics on evolutionary processes. Much of the differences among the conflicting evolutionary models for partitioning phenotypic variance can be accounted for by quantifying the distributions and phenotypic effects of epimutations in multiple different species (Kronholm and Collins 2016). Yet those variables are presently largely unknown and we must wait for the empirical data to parameterize those variables (sensu van der Graaf et al. 2015).
Epigenetic effects on phenotypic variance
Quantitative genetic studies are based on understanding components of variance within the framework VP = VG + VE, where VP is the total phenotypic variance in a trait in a population, VG is the phenotypic variance attributed to genetic variance, and VE refers to all other sources of phenotypic variance that are not the genetic variance (Falconer and Mackay 1996). In this simple, dichotomous partitioning, VE is often misleadingly termed the “environmental variance”, even though it contains more than just environmental variance. But VE can also be defined sensu stricto as the phenotypic variance attributed only to environmental variance (Scheiner and Goodnight 1984). When the definition of VE is limited to the phenotypic variance attributed to truly environmental variance, two additional terms are partitioned out in the phenotypic variance equation: VGxE, the phenotypic variance attributed to genotype-by-environment interaction, and Vɛ, the residual or error variance that is not explained by any of the other sources of variation (i.e., not explained by VG, VE, or VGxE) (Scheiner and Goodnight 1984). We argue that each component of the equation VP = VG + VE + VGxE + Vɛ can be influenced by epigenetics, and an additional partition, COVGE, can also be influenced by epigenetics. We also show how each of the variance components from the expanded phenotypic variance equation can influence evolution. Therefore, the influences of epigenetics on any of the variance components cannot be safely set aside and ignored when thinking about evolutionary processes and evolutionary outcomes.
Epigenetics, V G, and evolution
The genetic variance portion of phenotypic variance, VG, can be dissected into: VA + VD + VI + VGɛ, where VA is the additive genetic variance, meaning the portion of the total genetic variance that is inherited in an additive fashion. VD (dominance variance), VI (gene–gene interaction), and VGɛ (residual genetic variance) are other portions of the total genetic variance (Falconer and Mackay 1996). VA is the portion of phenotypic variance that is necessary for evolutionary change, and when standardized by the total phenotypic variance (VP) is defined as the narrow sense heritability (h2). Narrow-sense heritability is important in both agricultural breeding and in evolutionary theory, because it describes variation that is necessary for traits to evolve (Lopez-Fanjul and Garcia-Dorado 2011). The importance of narrow-sense heritability to evolution is represented by the “breeder’s equation”, R = h2S, where R is the response to selection on a trait, h2 is the heritability of the trait, and S is the strength of selection on the trait (Falconer and Mackay 1996). In this equation, evolution is the response to selection.
To the extent that epigenetic mechanisms contribute additively to the phenotype, then epigenetic variance may be found in the additive genetic variance component, VA, of the total genetic variance. It may be possible to partition epigenetic variance from VA (see the section below titled, Incorporating epigenetic effects into quantitative genetics studies; Spencer 2002; Santure and Spencer 2006, 2011; Tal et al. 2010; Macdonald 2012; Varona et al. 2015). But epigenetic mechanisms can also contribute to dominance deviations, because there is no reason to assume that epigenetic re-patterning of chromosomes should have purely additive effects on the phenotype. Therefore, epigenetics can also contribute to the variance in dominance deviations (VD) portion of the total genetic variance. The contribution of epigenetics to dominance deviations has not been thoroughly investigated, to our knowledge, but it would helpful to learn more about how much epigenetic mechanisms contribute to VD.
Epigenetically re-patterned genes may influence the expression of other genes and their effects on the phenotype, and thus epigenetics can influence the epistatic variance, VI, portion of the total genetic variance as well. Shivaprasad et al. (2012) studied gene expression patterns in the F1 progeny of a cross between cultivated tomato (Solanum lycopersicum) and a wild relative (Solanum pennellii) and found that micro or small interfering (si)RNAs were more abundant in hybrids than in either parent, and that accumulation of such transgressive sRNAs correlated with suppression of the corresponding target genes. In one instance, this effect was associated with hypermethylation of the corresponding genomic DNA. Smith and Weigel (2012) pointed out that many sRNAs are produced from non-coding regions or transposable elements, which diverge more quickly than protein-coding genes and thus provide more opportunity for unexpected genetic interactions. To the extent that gene expression changes are mediated epistatically by epigenetic changes (as in Shivaprasad et al. 2012), epigenetics can be influencing VI.
Epigenetic effects on VG can most obviously influence evolution via their effects on VA (the additive component of the total genetic variance), since the response to selection (evolution) in the breeder’s equation depends, in part, on h2, which is a standardized measure of VA. But epigenetic effects on VI (the epistatic component of the total genetic variance) can also be converted to additive genetic variance by the action of genetic drift and genetic bottlenecks that cause some loci to become fixed, eliminating the interaction effects among loci (Cheverud and Routman 1995; Hill 2017). The epigenetic effects that may be involved in epistatic interactions could be evolutionarily important during speciation events, when founders are isolated into smaller populations and genetic diversity is reduced. Epigenetic variation is reprogrammed through hybridization among species in the wild and in agricultural contexts (Salmon et al. 2005; Salmon et al. 2008), and may strengthen reproductive barriers among species (Smith and Weigel 2012). In summary, epigenetic influences on VG can influence evolution, via their effects on VI as well as via their effects on VA; the nonadditive component of VG should not be ignored when accounting for epigenetic influences on phenotypic variance and evolution.
Epigenetics, V E sensu stricto, and evolution
Phenotypic plasticity is the ability of a genotype to produce distinct phenotypes in response to different environmental conditions (Pigliucci 2001). It can be quantified for a single individual, or for multiple replicates of the same genotype, across different environments. A “genotype” in this instance can refer to individuals with specific configurations of alleles at a locus. It can also refer to clonal copies of an individual (e.g., Fallopia spp.; Richards et al. 2008; Parepa et al. 2014), apomictic offspring (e.g., Taraxicum; Verhoeven and van Gurp 2012) or an inbred strain of a species such as the Landsberg erecta strain of Arabidopsis thaliana (Koornneef and Meinke 2010) or the Sprague–Dawley strain of the rat Rattus norvegicus (Hsu and Lai 2007). Plasticity is typically measured by the phenotype in one environment minus the phenotype in the other environment, and is visualized on a graph as a “norm of reaction” (Pigliucci 2001; Fig. 1). The greater the difference in the phenotype between the two environments, the greater the plasticity. Phenotypic plasticity can also be described as the phenotypic variance in a dataset that is due to differences in the phenotype among two or more different environments. Understood in this way, plasticity is represented by the term VE sensu stricto in the partitioning of phenotypic variance (Scheiner and Goodnight 1984).

The phenotype (y-axis) of the same individual/inbred strain/genotype in two different environments (x-axis) as a norm of reaction. The phenotype in Environment 2 minus the phenotype in Environment 1 indicates how much phenotypic plasticity (VE) there is
To the extent that changes in the phenotype in different environments arise from changes in gene expression, epigenetic modifications could be contributing to those changes in gene expression (Duncan et al. 2014). There are examples of epigenetically-mediated plasticity in many taxa, including flowering plants (Geng et al. 2013; Zhang et al. 2013; Herman and Sultan 2016), yeast (Herrera et al. 2012), honeybees (Kucharski et al. 2008), ants (Bonasio et al. 2012; Simola et al. 2013), mammals (Oberlander et al. 2008; Godfrey et al. 2011; Lim et al. 2012; Hunter et al. 2015), and birds (Lindqvist et al. 2007; Natt et al. 2009; Goerlich et al. 2012; Nätt et al. 2012). Generally speaking, epigenetic modifications are likely a common mechanism of phenotypic plasticity (Nicotra et al. 2010; Richards et al. 2010; Geng et al. 2013; Herman et al. 2014; Herman and Sultan 2016), and can account for some of the phenotypic variance via their effects on response to environment (VE).
A relevant example of phenotypic plasticity mediated by epigenetic alterations comes from Thorson et al. (2017). They examined morphological divergence and epigenetic variation in asexual freshwater snail populations of the species Potamopyrgus antipodarum from contrasting lake and river habitats. They found that populations exhibit habitat specific differences in shell shape that are adaptive given the water current speeds of their environments. These differences were associated with significant genome-wide DNA methylation differences among snails from different habitats, and the different methylation patterns were repeated in replicate habitats. Because these snails contain few molecular genetic differences, Thorson et al. (2017) hypothesize that the differences in the shell morphology among habitats are due to adaptive phenotypic plasticity that is mediated by epigenetic mechanisms, specifically DNA methylation throughout the genome. It is possible that the observed differences in methylation among habitats are not just environmentally induced but also heritable, but this additional possibility was not tested.
While epigenetic modifications may provide insight into the molecular details of phenotypic plasticity (Nicotra et al. 2010), the two are not entirely interchangeable. As described above, epigenetics is also found in the VG part of the dissection of phenotypic variance, which is not a plastic response to environment. Furthermore, phenotypic plasticity can be due to many biochemical mechanisms (reviewed in Kelly et al. 2012), and not all of these mechanisms involve epigenetics. For instance, phenotypic plasticity can be due to the kinetic constraints of enzyme functions in response to temperature. This type of plasticity can be seen in plants, which have optimal temperature ranges for photosynthesis, driven by the kinetic properties of the enzymes and other mechanisms involved (Yamasaki et al. 2002). Therefore, photosynthetic reactions can go slower or faster depending on the temperature regime experienced by the plant, and enhanced growth of a plant under moderately higher temperatures could be due to increased rates of enzymatic reactions and not necessarily to epigenetic mechanisms. Optimal environmental ranges for enzymes are a fundamental aspect of cellular physiology across the domains of life (Geueke and Kohler 2010), so this example is generalizable to any taxon. Similarly, to the extent that an organism grows larger in response to nutrients simply because it has more materials to build more cells and more structures, this does not necessarily result from an epigenetic mechanism mediating the plastic response of organism size to nutrient levels. In summary, some phenotypic variance can be attributed to epigenetic modifications that are not plasticity (like epigenetics influencing VG) and some phenotypic variance is due to plasticity that is not the result of epigenetic modifications.
Epigenetic effects on VE sensu stricto may not have obvious ramifications for evolution, given that environmental variation does not have a heritable component to it. But epigenetic states are different from DNA sequence variation in that epigenetic states can be environmentally induced and then inherited in the germ line (Morgan et al. 1999; Waterland and Jirtle 2003; Verhoeven et al. 2010; Richards et al. 2012; Frésard et al. 2013; and reviewed in Richards et al. 2017). Thus, epigenetic mechanisms can account for some of the plastic response of a trait to the environment (which is VE sensu stricto) that is then converted into additive genetic variance. Surprising as it may first seem, epigenetic influences on VE sensu stricto have evolutionary implications for the calculation of VA, and therefore epigenetic influences on VE sensu stricto may potentially influence evolution.
Epigenetics, V GxE, and evolution
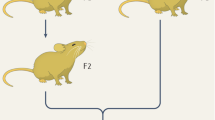
The effects of the environment on phenotype can vary by genotype (Fig. 2). In other words, the expression and function of genes can change depending on the specific alleles comprising a genotype (Gillespie and Turelli 1989). This widespread phenomenon (Baye et al. 2011; De Marais et al. 2013; Rauw and Gomez-Raya 2015), called the “genotype-by-environment interaction”, is partitioned from the total phenotypic variance (Scheiner and Goodnight 1984). Any genotypes in Fig. 2 that have non-zero slopes are displaying plasticity (Pigliucci 2001), and the fact that different genotypes have different slopes indicates that there is genetic variation for plasticity (Scheiner and Lyman 1989). Thus, there are environmental and genetic components in VGxE.

The phenotype (y-axis) of three different individuals/inbred strains/genotypes in two different environments (x-axis) as a norm of reaction. Each individual/inbred strain/genotype is indicated by a different symbol + color. The fact that the three genotypes have different values for the phenotype in Environment 2 minus the phenotype in Environment 1 indicates that there is genetic variation for plasticity (VGxE)
Just as there can be epigenetic contributions to the environmental (VE) and genetic (VG) components of phenotypic variance, there can also be epigenetic contributions to the genotype-by-environment interaction (VGxE). Take, for instance, the genotypes in Fig. 2 in environment 1. Within that environment, there is genetic variance in the phenotype—the three genotypes have different genotypic means. As described above, epigenetics can be contributing to the genetic variance. Similarly, take the same genotypes in Fig. 2 in environment 2. Within that environment, there is also genetic variation in the phenotype and, therefore, epigenetic mechanisms can be contributing to the genetic variance within that environment. Now take genotype 1 and examine how its phenotype changes across environments. This is phenotypic plasticity, and epigenetic mechanisms can contribute to phenotypic plasticity, as described above.
The signature of epigenetic effects on VGxE has been documented in a few studies. Bossdorf et al. (2010) treated a set of natural inbred lines of Arabidopsis thaliana with the demethylating agent 5-azacytidine and examined the consequences of this treatment for plasticity of plant traits to nutrient levels. By looking at how demethylation influenced the plastic responses of the inbred lines as compared to their control counterparts, they found that the effect of epigenetic re-patterning on plastic responses varies among inbred lines (Fig. 3). But the changes in the plastic responses only weakly matched what would be expected by the relatedness of the lines to one another. This suggests that the inbred lines differed in their methylation patterns in ways that are at least partly decoupled from their DNA sequence similarity. Similarly, Tatra et al. (2000) investigated the response of DNA methylation to irradiance-mediated plasticity of stem elongation in two ecotypes of Stellaria longipes, using a multifactorial design of light and 5-azacytidine. They found that the plastic responses to light were altered differently by demethylation among the two different ecotypes. In another study, Herman and Sultan (2016) used demethylation in Polygonum persicaria to show that the inheritance of adaptive response in offspring of drought-exposed plants was mediated by DNA methylation. The effect of demethylation on inheritance of the adaptive response varied among inbred lines. These studies demonstrate empirically that epigenetic modifications can account for some of the differences in plastic responses among genotypes. To the extent that epigenetic differences are inherited, epigenetic mechanisms may account for some of the inherited differences in plasticity among different genotypes.

Genotypic variation in the effects of experimental demethylation on plant phenotypic plasticity. The reaction norms illustrate how treatment with 5-azacytidine alters the phenological response to nutrient additions in 22 genotypes of Arabidopsis thaliana. Two genotypes with contrasting responses are highlighted in bold. Reproduced from Bossdorf et al. (2010) with permission
The genotype-by-environment interaction can be thought of as genetic variance for plasticity, and can be broken down into additive and dominance variance components. This allows for calculating the heritability (in the narrow-sense) of plasticity, which can evolve (Scheiner and Lyman 1989), There are many examples of the evolution of plastic responses to the environment (Dudley and Schmitt 1995; Gotthard and Nylin 1995; Pigliucci 2001). As such, epigenetic influences on VGxE can influence evolution, because VGxE contains heritable genetic variation for plasticity and some of this heritable variation can be due to epigenetic mechanisms.
Another way in which VGxE can influence evolution is through genetic assimilation. Genetic assimilation is the process where a phenotype originally produced in response to an environmental stimulus later becomes genetically encoded via selection, and the plastic response is lost, due to a directional shift in the environment (Waddington 1956; Pigliucci and Murren 2003; Crispo 2007). Phenotypic plasticity can thus can serve as a bridge for populations to adapt to changing environments, and evolution via genetic assimilation can later developmentally hard-wire the induced plastic responses. Under a scenario of genetic assimilation, selection first acts on genetic variation for plasticity (VGxE), favoring individuals that are able to alter their phenotype in ways that better match the new environment; if the ancestral environment is later lost, selection will subsequently favor constitutive induction of the novel phenotype, due to costs associated with maintaining the metabolic/developmental machinery for a plastic response to the environment (Pigliucci and Murren 2003). To the extent that epigenetic variation among individuals account for some of the VGxE in response to a novel environment, epigenetic mechanisms provide some of the genetic variance in plasticity that selection can act on in the new environment. This plasticity can later become genetically encoded and constitutive via evolution by genetic assimilation.
Epigenetics, COVGE, and evolution
When the occurrence of specific environmental conditions depends on an organism’s genotype, or vice versa, there is a genotype–environment covariance (COVGE; Falconer and Mackay 1996). The genotype–environment covariance combines the genetic variance together with the environmental variance (Falconer and Mackay 1996). A standardized version of this covariance is often presented in the literature as a genotype–environment correlation (rGE; Jaffee and Price 2007). Because epigenetic mechanisms can play a role in genetic variance, epigenetic mechanisms can also play a role in COVGE, which is based, in part, on genetic variance. As with each of the other components of the phenotypic variance formula, the epigenetic contribution to COVGE must be accounted for, either by cross-factorial experimentation where each genotype experiences each environment equally, or by explicitly estimating COVGE as part of partitioning of phenotypic variance. If COVGE is not accounted for, then the phenotypic variance it introduces can confound estimates of VG or VGxE (Falconer and Mackay 1996; Jaffee and Price 2007). A covariance between genotype and environment is particularly likely to arise when considering epigenetic mechanisms of phenotypic variance, because epigenetic changes can be environmentally induced and then inherited (Morgan et al. 1999; Waterland and Jirtle 2003; Verhoeven et al. 2010; Richards et al. 2012; Frésard et al. 2013; and reviewed in Richards et al. 2017). Given that organisms tend to inherit their environments from their parents (Oyama et al. 2001), the environmental induction of epigenotypes, combined with the inheritance of parental environments, means that the prevalence of different epigenotypes should be particularly nonrandom with respect to the environments in which the epigenotypes are found. Without the COVGE term in the phenotypic variance formula, the mathematical assumption is that the (epi)genetic variance and environmental variance are unrelated, so the addition of this term is important for properly partitioning phenotypic variance when the asusmption of (epi)gentype-environment independence is violated.
Incorrect estimation of VG and VGxE could lead to an incorrect heritability estimate of a trait or trait plasticity, and the response of a trait to selection (i.e., evolution) could be miscalculated. The genotype–environment covariance is often ignored in quantitative genetic studies, because it is assumed to be of negligible importance (Falconer and Mackay 1996). However, COVGE could be a large portion of the total phenotypic variance when epigenetic effects are involved, due to induction of epigenetic changes by the environment and the subsequent inheritance of those epigenetic changes (Bonduriansky and Day 2009; Slatkin 2009; Geoghegan and Spencer 2012, 2013; Richards et al. 2017). We propose that accounting for the epigenotype–environment covariance could be accomplished by first clustering individuals, based on their molecular epigenetic marker similarities (using a Bayesian clustering algorithm for example; Pritchard et al. 2000), and then calculating the epigenotype–environmental covariance by combining the information about the environments that the individuals experienced, the individuals’ phenotypes, and the epigenetic clusters that the individuals were assigned to. Development of this or similar approaches is beyond the scope of this paper, but methods for parsing out the epigenotype–environment covariance are needed to properly parse phenotypic variance into its various constituents. It should be possible to calculate the epigenetic contribution to COVGE, but the methods for doing so have not yet been fleshed out.
Epigenetics, V ɛ, and evolution
The residual or error variance, Vɛ, as defined by Scheiner and Goodnight (1984), includes only measurement error, microenvironmental variance, and developmental stochasticity. Microenvironmental variance refers to environmental variance that was not explicitly accounted for in the study design. If there are a large of amount of environmental effects that are not explicitly accounted for by VE sensu stricto in the study design, then the microenvironmental variance can actually be larger than VE sensu stricto (Mulder et al. 2013). Yet the microenvironmental variance is an invisible constituent of Vɛ in the phenotypic variance partitioning, because by definition it was not measured. Given that epigenetic changes can influence the environmental variance of the phenotype, as described above, epigenetic changes can also influence microenvironmental variance of the phenotype, because microenvironmental variance is just phenotypic variance attributable to environmental axes that were unmeasured. This also means that epigenetic effects on microenvironmental variance can influence evolution in the same was that epigenetic effects on environmental variance can influence evolution, as described above.
Methods for incorporating epigenetic effects into quantitative genetics studies
With the growing evidence that epigenetic changes can have different ramifications for evolution than DNA sequence changes, methods are being developed to actually incorporate epigenetic effects into models of quantitative genetics. Models of genomic imprinting have found that the estimation of the total genetic variance is not affected by imprinting, but the partitioning of the genetic variance in the phenotype changes and the response to selection for a highly imprinted trait cannot be predicted by the breeder’s equation (Spencer 2002; Santure and Spencer 2006, 2011). Day and Bonduriansky (2011) propose a model for non-genetic inheritance more generally based on the Price equation (Price 1972) that emphasizes the interactions between genetic and epigenetic effects with the following parameters for both (1) the effects of selection, (2) changes that occur in transmission from the parent to offspring generation (“reproductive transmission”) and (3) changes that occur in the parent generation (“survival transmission”). This model allows for changes in DNA sequence, the fact that some genomic contexts are more likely to acquire epigenetic effects than others, and the change in the epigenetic component is dependent on genotype. They found that epigenetic variation can determine the pattern of phenotypic variation and that phenotypic change can be decoupled from the dynamics of the genotype. Slatkin (2009) modeled disease risk as a function of diallelic genetic and epigenetic loci, and considered that for every individual offspring per generation, there is an opportunity for any individual epigenetic state to be reset. Therefore, not all of the epigenetic differences will persist in the offspring, and the probability that relatives resemble each other in epigenetic state is less than expected by their relatedness. Slatkin (2009) concluded that epigenetic effects do not contribute substantially to the heritability of diseases, but Slatkin cautioned that his conclusions depended heavily on the persistence times of heritable epialleles. How persistent epialleles are across various taxa is currently an unanswered question.
While several models that incorporate epigenetic changes into quantitative genetic studies require the estimation of molecular epigenetic processes, and particularly the rate of gain and loss of epigenetic effects across the genome, a model developed by Tal et al. (2010) proposes the use of only phenotypic datasets combined with a detailed pedigree. The Tal et al. (2010) model considers transmissibility (the probability of transmission of the epigenetic state) and environmental induction (the probability of the epigenetic state being induced) by adding a term, VC, to the equation VP = VG + VE, that reapportions some of the phenotypic variance from VG and VE. Calculating VC requires determining the phenotypic covariances between parents and offspring, between sibs, and between uncles and nephews. The design allows for estimation of the heritable epigenetic variance and epigenetic transmissibility using information about the number of opportunities for epigenetic reset between generations, and assumptions about environmental induction. They introduce a new term, γ2, which is the epigenetic heritability, or the proportion of phenotypic variance attributable to potentially heritable epigenetic variance. Different from standard heritability (h2), the contribution of epigenetic heritability (γ2) to parent-offspring similarity depends on the transmissibility coefficient: if the epigenetic states tend to be reset frequently, then the impact of γ2 on parent-offspring similarity is low; if the epigenetic states tend to be transgenerationally stable, then the impact of γ2 on parent-offspring similarity is high. The introduction of a new type of heritability with different ramifications for similarity among relatives could enhance our understanding of evolutionary processes by describing how different components of heritable phenotypic variation respond to selection.
The Tal et al. (2010) study provides a practical way forward for parsing out epigenetic influences in quantitative genetic studies. But like in other studies, Tal et al. (2010) do not account for the influence of epigenetic differences on VGxE. Perhaps the Tal et al. (2010) formula could be reformulated to account for epigenetic influences on VGxE, by partitioning VCxE from VGxE in a similar manner to what they describe for partitioning VC from VG and VE (sensu lato). The development of such a method lies outside the scope of this paper, but it would be a fruitful area of future research. Similarly, Richards et al. (2017) recommend that future studies on phenotypic variance should parse out a higher-order interaction term involving epigenetic effects, which could be something like VGxExC. This variance component would account for the ways in which DNA-encoded differences in plasticity change depending on the epigenetic effects.
It is important to note that Tal et al. (2010) define epigenetic inheritance broadly to include the transmission of phenotypic variation by any means besides transmission of DNA sequence variation. Their definition of epigenetics is broader than the more mechanistic, cellular definition of epigenetics that we have described (namely, chemical modifications of chromatin or transcribed DNA that can influence gene activity and expression without changes in DNA sequence; Jablonka and Raz 2009; Kilvitis et al. 2014), and their definition includes additional parental effects (Kirkpatrick and Lande 1989; Wolf and Wade 2016). It may be that there is no way in principle to separate epigenetics effects sensu stricto from other non-genetic forms of inheritance using a purely phenotypic dataset and a pedigree. Yet, despite potentially overestimating the epigenetic effects sensu stricto, the Tal et al. (2010) approach can be used to set upper bounds on the amount epigenetic influence on phenotypic variance and provide data to generate hypotheses for future research. For instance, they give the example of rapidly increasing heritability in inbred lines (Grewal 1962; Hoi-Sen 1972; Lande 1975), where one may expect VA (and therefore h2) to be small due to a lack of DNA sequence diversity. The classic interpretation is that the inbred lines must have a DNA mutation rate that is three orders of magnitude higher than is normally expected in order to increase the heritability and increase it that quickly. But if the estimates of VC, γ2, and the transmissibility coefficient are greater than zero, then the heritability may be partially generated by epigenetic mechanisms. The specific epigenetic mechanisms that contribute to heritability in the inbred lines could be examined in follow-up studies that use epigenomic mapping techniques (e.g., Zhang et al. 2013; Cortijo et al. 2014; Kooke and Keurentjes 2015). Overall, the Tal et al. (2010) study provides a pragmatic approach for teasing out epigenetic influences on phenotypic variance and heritability, but the methods developed so far are imperfect and require further development to accurately quantify epigenetic influences.
Conclusions
The modern era of epigenetics research presents opportunities for advancing our understanding of evolutionary processes, just as the genomics era more generally has greatly influenced evolutionary disciplines. We have demonstrated how epigenetic phenomena can contribute to each of the components of phenotypic variance in the expanded formula: VG, VE, VGxE, Vɛ, and COVGE. It is important to consider the influence of epigenetic phenomena on phenotypic variance when hypothesizing about the mechanisms behind phenotypic variance observed in a quantitative genetic study. One cannot assume that only DNA sequence-based phenomena account for patterns when epigenetic and other non-genetic phenomena could be playing a role as well; and accounting for parental effects will not necessarily capture epigenetic phenomena. The contribution of epigenetic mechanisms cannot be easily cast aside, especially given the increasing reliance on molecular mechanisms as a part of our understanding of evolutionary processes, and the fact that epigenetic mechanisms can have different ramifications for evolution from DNA sequence-based mechanisms. Some existing studies have developed methods for partitioning epigenetic variance from phenotypic variance, which represents a step in the right direction, but we need more methods that carefully consider all of the areas where variance due to epigenetic factors may need to be accounted for. The challenge of developing methods to parse out all of the ways that epigenetics can contribute to evolutionary models will require significant effort, but the difficulty of deciphering the role of epigenetic mechanisms does not preclude its importance.
References
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17:487
Baye TM, Abebe T, Wilke RA (2011) Genotype–environment interactions and their translational implications. Per Med 8(1):59–70
Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K et al. (2011) Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480:245
Bell JT, Spector TD (2011) A twin approach to unraveling epigenetics. Trends Genet 27(3):116–125
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009) An operational definition of epigenetics. Gene Dev 23(7):781–783
Bonasio R, Li Q, Lian J, Mutti NS, Jin L, Zhao H et al. (2012) Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol 22(19):1755–1764
Bonduriansky R, Day T (2009) Nongenetic inheritance and its evolutionary implications. Annu Rev Ecol Evol Syst 40:103–125
Bossdorf O, Arcuri D, Richards C, Pigliucci M (2010) Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evol Ecol 24(3):541–553
Brachi B, Morris GP, Borevitz JO (2011) Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol 12(10):232
Burggren W (2016) Epigenetic inheritance and its role in evolutionary biology: re-evaluation and new perspectives. Biology 5(2):24
Caballero A, Tenesa A, Keightley PD (2015) The nature of genetic variation for complex traits revealed by GWAS and regional heritability mapping analyses. Genetics 201(4):1601–1613
Chen F, Evans A, Gaskell E, Pham J, Tsai M-C (2011) Regulatory RNA: the new age. Mol Cell 43(6):851–852
Cheverud JM, Routman EJ (1995) Epistasis and its contribution to genetic variance components. Genetics 139(3):1455–1461
Cortijo S, Wardenaar R, Colome-Tatche M, Gilly A, Etcheverry M, Labadie K et al. (2014) Mapping the epigenetic basis of complex traits. Science 343(6175):1145–1148
Crispo E (2007) The Baldwin effect and genetic assimilation: revisiting two mechanisms of evolutionary change mediated by plasticity. Evolution 61(11):2469–2479
Cubas P, Vincent C, Coen E (1999) An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401(6749):157–161
Day T, Bonduriansky R (2011) A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am Nat 178:E18–E36
De Marais DL, Hernandez KM, Juenger TE (2013) Genotype-by-environment interaction and plasticity: exploring genomic responses of plants to the abiotic environment. Annu Rev Ecol Evol Syst 44:5–29
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB (2010) Rare variants create synthetic genome-wide associations. PLoS Biol 8(1):e1000294
Douhovnikoff V, Dodd RS (2015) Epigenetics: a potential mechanism for clonal plant success. Plant Ecol 216:227–233
Duncan EJ, Gluckman PD, Dearden PK (2014) Epigenetics, plasticity, and evolution: How do we link epigenetic change to phenotype? J Exp Zool B Mol Dev Evol 322(4):208–220
Dudley SA, Schmitt J (1995) Genetic differentiation in morphological responses to simulated foliage shade between populations of Impatiens capensis from open and woodland sites. Funct Ecol 9(4):655–666
Edwards SM, Sorensen IF, Sarup P, Mackay TF, Sorensen P (2016) Genomic prediction for quantitative traits is improved by mapping variants to gene ontology categories in Drosophila melanogaster. Genetics 203(4):1871–1883
Erickson DL, Fenster CB, Stenoien HK, Price D (2004) Quantitative trait locus analyses and the study of evolutionary process. Mol Ecol 13(9):2505–2522
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics. Prentice Hall, New York
Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science 330(6004):622–627
Frésard L, Morisson M, Brun J-M, Collin A, Pain B, Minvielle F et al. (2013) Epigenetics and phenotypic variability: Some interesting insights from birds. Genet Sel Evol 45(1):16
Fultz D, Choudury SG, Slotkin RK (2015) Silencing of active transposable elements in plants. Curr Opin Plant Biol 27:67–76
Furrow RE (2014) Epigenetic inheritance, epimutation, and the response to selection. PLoS One 9(7):e101559
Geoghegan JL, Spencer HG (2012) Population-epigenetic models of selection. Theor Popul Biol 81:232–242
Geoghegan JL, Spencer HG (2013) Exploring epiallele stability in a population-epigenetic model. Theor Popul Biol 83:136–144
Geng Y, Gao L, Yang J (2013) Epigenetic flexibility underlying phenotypic plasticity. In: Lüttge U, Beyschlag W, Francis D, Cushman J (eds) Progress in botany: vol. 74. Springer, Berlin, Heidelberg, p 153–163
Geueke B, Kohler H-PE (2010) Enzyme assays, substrate specificities, kinetic parameters: measurement of enzyme activities. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Heidelberg, p 4195–4202
Gibney ER, Nolan CM (2010) Epigenetics and gene expression. Heredity 105(1):4–13
Gillespie JH, Turelli M (1989) Genotype-environment interactions and the maintenence of polygenic variation. Genetics 121(1):129–138
Goddard ME, Whitelaw E (2014) The use of epigenetic phenomena for the improvement of sheep and cattle. Front Genet 5:247
Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C et al. (2011) Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 60(5):1528–1534
Goerlich VC, Natt D, Elfwing M, Macdonald B, Jensen P (2012) Transgenerational effects of early experience on behavioral, hormonal and gene expression responses to acute stress in the precocial chicken. Horm Behav 61(5):711–718
González-Recio O (2011) Epigenetics: a new challenge in the post-genomic era of livestock. Front Genet 2:106
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333
Gotthard K, Nylin S (1995) Adaptive plasticity and plasticity as an adaptation: a selective review of plasticity in animal morphology and life history. Oikos 74(1):3–17
Grewal MS (1962) The rate of genetic divergence of sublines in the C57BL strain of mice. Genet Res 3(2):226–237
Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA et al. (2013) Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339(6118):448
Hadfield J (2012) The quantitative genetic theory of parental effects. In: Royle NJ, Smiseth PT, Kölliker M (eds) The evolution of parental care. Oxford University Press, New York
Hemani G, Knott S, Haley C (2013) An evolutionary perspective on epistasis and the missing heritability. PLoS Genet 9(2):e1003295
Henderson IR, Jacobsen SE (2007) Epigenetic inheritance in plants. Nature 447(7143):418–424
Herman JJ, Spencer HG, Donohue K, Sultan SE (2014) How stable “should” epigenetic modifications be? Insights from adaptive plasticity and bet hedging. Evolution 68(3):632–643
Herman JJ, Sultan SE (2016) DNA methylation mediates genetic variation for adaptive transgenerational plasticity. Proc Biol Sci 283:20160988
Herrera CM, Medrano M, Bazaga P (2013) Epigenetic differentiation persists after male gametogenesis in natural populations of the perennial herb Helleborus foetidus (Ranunculaceae). PLoS One 8:e70730
Herrera CM, Pozo MI, Bazaga P (2012) Jack of all nectars, master of most: DNA methylation and the epigenetic basis of niche width in a flower-living yeast. Mol Ecol 21(11):2602–2616
Hill WG (2012) Quantitative genetics in the genomics era. Curr Genom 13(3):196–206
Hill WG (2017) “Conversion” of epistatic into additive genetic variance in finite populations and possible impact on long-term selection response. J Anim Breed Genet 134(3):196–201
Hoi-Sen Y (1972) Is subline differentiation a continuing process in inbred strains of mice? Genet Res 19(1):53–59
Hsu C-C, Lai S-C (2007) Matrix metalloproteinase-2, -9 and -13 are involved in fibronectin degradation of rat lung granulomatous fibrosis caused by Angiostrongylus cantonensis. Int J Exp Pathol 88(6):437–443
Huff JT, Zilberman D (2014) Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 156(6):1286–1297
Hunter RG, Gagnidze K, McEwen BS, Pfaff DW (2015) Stress and the dynamic genome: steroids, epigenetics, and the transposome. Proc Natl Acad Sci USA 112(22):6828–6833
Huxley J (1942) Evolution: The Modern Synthesis. Allen & Unwin, London
Ikeda Y, Nishimura T (2015) The role of DNA methylation in transposable element silencing and genomic imprinting. In: Pontes O, Jin H (eds) Nuclear functions in plant transcription, signaling and development. Springer, New York, NY, p 13–29
Jablonka E, Lamb MJ (1989) The inheritance of acquired epigenetic variations. J Theor Biol 139:69–83
Jablonka E, Lamb MJ, Avital E (1998) Lamarckian mechanisms in Darwinian evolution. Trends Ecol Evol 13:206–210
Jablonka E, Raz G (2009) Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol 84(2):131–176
Jaffee SR, Price TS (2007) Gene-environment correlations: a review of the evidence and implications for prevention of mental illness. Mol Psychiatry 12(5):432–442
Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N et al. (2009) Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 5(6):e1000530
Josephs EB, Stinchcombe JR, Wright SI (2017) What can genome‐wide association studies tell us about the evolutionary forces maintaining genetic variation for quantitative traits? New Phytol 214(1):21–33
Kalisz S, Kramer EM (2008) Variation and constraint in plant evolution and development. Heredity 100(2):171–177
Kelly SA, Panhuis TM, Stoehr AM (2012) Phenotypic plasticity: molecular mechanisms and adaptive significance. Compr Physiol 2(2):1417–1439
Kilvitis HJ, Alvarez M, Foust CM, Schrey AW, Robertson M, Richards CL (2014) Ecological epigenetics. In: Landry CR, AubinHorth N (eds) Ecological genomics: ecology and the evolution of genes and genomes, vol. 781. Springer, New York, p 191–210.
Kirkpatrick M, Lande R (1989) The evolution of maternal characters. Evolution 43(3):485–503
Klironomos FD, Berg J, Collins S (2013) How epigenetic mutations can affect genetic evolution: model and mechanism. Bioessays 35(6):571–578
Kooke R, Keurentjes JJB (2015) Epigenetic variation contributes to environmental adaptation of Arabidopsis thaliana. Plant Signal Behav 10(9):e1057368
Kooke R, Kruijer W, Bours R, Becker F, Kuhn A, van de Geest H et al. (2016) Genome-wide association mapping and genomic prediction elucidate the genetic architecture of morphological traits in Arabidopsis. Plant Physiol 170(4):2187
Koopaee HK, Koshkoiyeh AE (2014) SNPs genotyping technologies and their applications in farm animals breeding programs: review. Braz Arch Biol Technol 57:87–95
Koornneef M, Meinke D (2010) The development of Arabidopsis as a model plant. Plant J 61(6):909–921
Kronholm I, Collins S (2016) Epigenetic mutations can both help and hinder adaptive evolution. Mol Ecol 25(8):1856–1868
Kucharski R, Maleszka J, Foret S, Maleszka R (2008) Nutritional control of reproductive status in honeybees via DNA methylation. Science 319(5871):1827
Laland KN, Uller T, Feldman MW, Sterelny K, Muller GB, Moczek A et al. (2015) The extended evolutionary synthesis: its structure, assumptions and predictions. Proc R Soc Lond B Biol Sci 282(1813):20151019
Lande R (1975) The maintenance of genetic variability by mutation in a polygenic character with linked loci. Genet Res 26(3):221–235
Leimar O, McNamara JM (2015) The evolution of transgenerational integration of information in heterogeneous environments. Am Nat 185(3):E55–E69
Li X, Zhou Z, Ding J, Wu Y, Zhou B, Wang R et al. (2016) Combined linkage and association mapping reveals qtl and candidate genes for plant and ear height in maize. Front Plant Sci 7:833
Liebl AL, Schrey AW, Richards CL, Martin LB. (2013) Patterns of DNA methylation throughout a rangeexpansion of an introduced songbird. Integr Comp Biol 53(2):351–358
Liberles DA (2001) Evolution enters the genomic era. Genome Biol 2(11): reports4026.4021-reports4026.4025
Lim AL, Ng S, Leow SC, Choo R, Ito M, Chan YH et al. (2012) Epigenetic state and expression of imprinted genes in umbilical cord correlates with growth parameters in human pregnancy. J Med Genet 49(11):689–697
Lindqvist C, Janczak AM, Nätt D, Baranowska I, Lindqvist N, Wichman A et al. (2007) Transmission of stress-induced learning impairment and associated brain gene expression from parents to offspring in chickens. PLoS One 2(4):e364
Lopez-Fanjul C, Garcia-Dorado A (2011) The fuel of evolution. Heredity 106(4):535–536
Macdonald W (2012) Epigenetic mechanisms of genomic imprinting: common themes in the regulation of imprinted regions in mammals, plants, and insects. Genet Res Int 2012:1–17
Mackay TFC (2001) The genetic architecture of quantitative traits. Annu Rev Genet 35(1):303–339
Mackay TFC (2014) Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat Rev Genet 15:22
McNiven VTK, LeVasseur-Viens H, Kanippayoor RL, Laturney M, Moehring AJ (2011) The genetic basis of evolution, adaptation and speciation. Mol Ecol 20(24):5119–5122
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829
Migicovsky Z, Kovalchuk I (2011) Epigenetic memory in mammals. Front Genet 2:28
Morgan HD, Sutherland HG, Martin DI, Whitelaw E (1999) Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23(3):314–318
Mulder HA, Rönnegård L, Fikse WF, Veerkamp RF, Strandberg E (2013) Estimation of genetic variance for macro- and micro-environmental sensitivity using double hierarchical generalized linear models. Genet Sel Evol 45(1):23–23
Natt D, Lindqvist N, Stranneheim H, Lundeberg J, Torjesen PA, Jensen P (2009) Inheritance of acquired behaviour adaptations and brain gene expression in chickens. PLoS ONE 4(7):e6405
Nätt D, Rubin C-J, Wright D, Johnsson M, Beltéky J, Andersson L et al. (2012) Heritable genome-wide variation of gene expression and promoter methylation between wild and domesticated chickens. BMC Genom 13(1):59
Nicotra AB, Atkin OK, Bonser SP, Davidson AM, Finnegan EJ, Mathesius U et al. (2010) Plant phenotypic plasticity in a changing climate. Trends Plant Sci 15:684–692
Niederhuth CE, Bewick AJ, Ji L, Alabady MS, Kim KD, Li Q et al. (2016) Widespread natural variation of DNA methylation within angiosperms. Genome Biol 17(1):194
Niederhuth CE, Schmitz RJ (2017) Putting DNA methylation in context: from genomes to gene expression in plants. Biochim Biophys Acta 1860(1):149–156
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM (2008) Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3(2):97–106
Oyama S, Griffiths PE, Gray RD (2001) Cycles of contingency: developmental systems and evolution. MIT Press, Cambridge, MA
Pál C, Miklós I (1999) Epigenetic inheritance, genetic assimilation and speciation. J Theor Biol 200:19–37
Parepa M, Fischer M, Krebs C, Bossdorf O (2014) Hybridization increases invasive knotweed success. Evol Appl 7:413–420
Pigliucci M (2001) Phenotypic plasticity: beyond nature versus nurture. Johns Hopkins University Press, Baltimore
Pigliucci M (2007) Do we need an extended evolutionary synthesis? Evolution 61:2743–2749
Pigliucci M, Murren CJ (2003) Perspective: genetic assimilation and a possible evolutionary paradox: can macroevolution sometimes be so fast as to pass us by? Evolution 57(7):1455–1464
Pigliucci M, Müller GB (2010) Evolution, the extended synthesis. MIT Press, Cambridge, MA
Plutynski A (2009) The modern synthesis. Taylor and Francis. https://www.rep.routledge.com/articles/thematic/modern-synthesis-the/v-1. Accessed 19 June 2018
Price GR (1972) Fisher’s fundamental theorem made clear. Ann Hum Genet 36(2):129–140
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67(1):170–181
Rapp RA, Wendel JF (2005) Epigenetics and plant evolution. New Phytol 168(1):81–91
Rauw WM, Gomez-Raya L (2015) Genotype by environment interaction and breeding for robustness in livestock. Front Genet 6:310
Rendina González AP, Chrtek J, Dobrev PI, Dumalasová V, Fehrer J, Mráz P et al. (2016) Stress-induced memory alters growth of clonal off spring of white clover (Trifolium repens). Am J Bot 103(9):1567–1574
Richards CL, Alonso C, Becker C, Bossdorf O, Bucher E, Colome-Tatche M et al. (2017) Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol Lett 20(12):1576–1590
Richards CL, Bossdorf O, Verhoeven KJF (2010) Understanding natural epigenetic variation. New Phytol 187(3):562–564
Richards CL, Schrey AW, Pigliucci M (2012) Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol Lett 15(9):1016–1025
Richards CL, Walls RL, Bailey JP, Parameswaran R, George T, Pigliucci M (2008) Plasticity in salt tolerance traits allows for invasion of novel habitat by Japanese knotweed s. l. (Fallopia japonica and F. x bohemica, Polygonaceae). Am J Bot 95(8):931–942
Robertson M, Richards C (2015) Non-genetic inheritance in evolutionary theory—the importance of plant studies. Non-Genet Inherit 2:3–11
Rockman MV (2012) The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 66(1):1–17
Roff DA (2007) A centennial celebration for quantitative genetics. Evolution 61(5):1017–1032
Ruppell O, Pankiw T, Page Jr. RE (2004) Pleiotropy, epistasis and new QTL: the genetic architecture of honey bee foraging behavior. J Hered 95(6):481–491
Salmon A, Ainouche ML, Wendel JF (2005) Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol Ecol 14(4):1163–1175
Salmon A, Clotault J, Jenczewski E, Chable V, Manzanares-Dauleux MJ (2008) Brassica oleracea displays a high level of DNA methylation polymorphism. Plant Sci 174(1):61–70
Sano H, Kim H-J (2013) Transgenerational epigenetic inheritance in plants. In: Grafi G, Ohad N (eds) Epigenetic memory and control in plants. Springer, Berlin, Heidelberg, p 233–253
Santure AW, Spencer HG (2006) Influence of mom and dad: quantitative genetic models for maternal effects and genomic imprinting. Genetics 173(4):2297–2316
Santure AW, Spencer HG (2011) Quantitative genetics of genomic imprinting: a comparison of simple variance derivations, the effects of inbreeding, and response to selection. G3 1(2):131–142
Saze H, Scheid OM, Paszkowski J (2003) Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34(1):65–69
Scheiner SM, Goodnight CJ (1984) The comparison of phenotypic plasticity and genetic variation in populations of the grass Danthonia spicata. Evolution 38(4):845–855
Scheiner SM, Lyman RF (1989) The genetics of phenotypic plasticity I. Heritability. J Evol Biol 2(2):95–107
Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O et al. (2013) Patterns of population epigenomic diversity. Nature 495(7440):193–198
Shea N, Pen I, Uller T (2011) Three epigenetic information channels and their different roles in evolution. J Evol Biol 24(6):1178–1187
Shivaprasad PV, Dunn RM, Santos BA, Bassett A, Baulcombe DC (2012) Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J 31(2):257–266
Simola DF, Ye C, Mutti NS, Dolezal K, Bonasio R, Liebig J et al. (2013) A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res 23(3):486–496
Slatkin M (2009) Epigenetic inheritance and the missing heritability problem. Genetics 182(3):845–850
Smith LM, Weigel D (2012) On epigenetics and epistasis: hybrids and their non-additive interactions. EMBO J 31(2):249–250
Spencer HG (2002) The correlation between relatives on the supposition of genomic imprinting. Genetics 161(1):411–417
Spens AE, Douhovnikoff V (2016) Epigenetic variation within Phragmites australis among lineages, genotypes, and ramets. Biol Invasions 18:2457
Stinchcombe JR, Hoekstra HE (2008) Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100:158
Tal O, Kisdi E, Jablonka E (2010) Epigenetic contribution to covariance between relatives. Genetics 184(4):1037
Tatra GS, Miranda J, Chinnappa CC, Reid DM (2000) Effect of light quality and 5-azacytidine on genomic methylation and stem elongation in two ecotypes of Stellaria longipes. Physiol Plant 109(3):313–321
Thorson JLM, Smithson M, Beck D, Sadler-Riggleman I, Nilsson E, Dybdahl M et al. (2017) Epigenetics and adaptive phenotypic variation between habitats in an asexual snail. Sci Rep 7(1):14139
Triantaphyllopoulos KA, Ikonomopoulos I, Bannister AJ (2016) Epigenetics and inheritance of phenotype variation in livestock. Epigenetics Chromatin 9:31
Uller T, English S, Pen I (2015) When is incomplete epigenetic resetting in germ cells favoured by natural selection? Proc R Soc Lond B Biol Sci 282(1811):20150682
van der Graaf A, Wardenaar R, Neumann DA, Taudt A, Shaw RG, Jansen RC et al. (2015) Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc Natl Acad Sci USA 112(21):6676–6681
Varona L, Munilla S, Mouresan EF, González-Rodríguez A, Moreno C, Altarriba J (2015) A Bayesian model for the analysis of transgenerational epigenetic variation. G3 5(4):477–485
Vergeer P, Wagemaker NC, Ouborg NJ (2012) Evidence for an epigenetic role in inbreeding depression. Biol Lett 8(5):798–801
Verhoeven KJ, Jansen JJ, van Dijk PJ, Biere A (2010) Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol 185(4):1108–1118
Verhoeven KJF, van Gurp TP (2012) Transgenerational effects of stress exposure on offspring phenotypes in apomictic dandelion. PLoS One 7(6):e38605
Verhoeven KJF, Preite V (2014) Epigenetic variation in asexually reproducing organisms. Evolution 68:644–655
Vongs A, Kakutani T, Martienssen RA, Richards EJ (1993) Arabidopsis thaliana DNA methylation mutants. Science 260(5116):1926–1928
Waddington CH (1942) Canalization of development and the inheritance of acquired characters. Nature 150:563–565
Waddington CH (1953) Genetic assimilation of an acquired character. Evolution 7:118–126
Waddington CH (1956) Genetic assimilation of the biothorax phenotype. Evolution 10(1):1–13
Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23(15):5293–5300
West-Eberhard MJ (2005) Developmental plasticity and the origin of species differences. Proc Natl Acad Sci USA 102:6543–6549
Wolf JB, Leamy LJ, Routman EJ, Cheverud JM (2005) Epistatic pleiotropy and the genetic architecture of covariation within early and late-developing skull trait complexes in mice. Genetics 171(2):683
Wolf JB, Wade MJ (2016) Evolutionary genetics of maternal effects. Evolution 70(4):827–839
Yamasaki T, Yamakawa T, Yamane Y, Koike H, Satoh K, Katoh S (2002) Temperature acclimation of photosynthesis and related changes in Photosystem II electron transport in winter wheat. Plant Physiol 128(3):1087–1097
Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH et al. (2015) Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet 47(10):1114–1120
Zhang Y-Y, Fischer M, Colot V, Bossdorf O (2013) Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol 197(1):314–322
Acknowledgements
We would like to thank the Society for the Study of Evolution and Sociedade Brasileira de Genética for funding our symposium, “Epigenetics and Evolutionary Processes”, at the Evolution annual meeting in Guarujá, Brazil in June 2015, where we refined this idea. This work was supported by funding from the National Science Foundation (USA) through a Research Opportunity Award (ROA) supplement to DEB-1419960 (CLR) to support JAB, and through IOS-1556820 (CLR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Banta, J.A., Richards, C.L. Quantitative epigenetics and evolution. Heredity 121, 210–224 (2018). https://doi.org/10.1038/s41437-018-0114-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0114-x
This article is cited by
-
Prenatal Stress Induces Long-Term Behavioral Sex-Dependent Changes in Rats Offspring: the Role of the HPA Axis and Epigenetics
Molecular Neurobiology (2023)
-
On the Usefulness of Behavior Genetics: Using Family Studies in Evolutionary Psychological Science to Improve Causal Inference and Sharpen Theory
Adaptive Human Behavior and Physiology (2023)
-
Cell-to-cell variability in inducible Caspase9-mediated cell death
Cell Death & Disease (2022)
-
Regional differences in clonal Japanese knotweed revealed by chemometrics-linked attenuated total reflection Fourier-transform infrared spectroscopy
BMC Plant Biology (2021)
-
Conservation and trans-regulation of histone modification in the A and B subgenomes of polyploid wheat during domestication and ploidy transition
BMC Biology (2021)
