
Abstract
Sheep, the Jaagsiekte sheep retrovirus (JSRV) and its endogenous forms (enJSRVs) are a good model to study long-time relationships between retroviruses and their hosts. Taking advantage of 76 whole genome resequencing data of wild and domestic Ovis, we investigated the evolution of this relationship. An innovative analysis of re-sequencing data allowed characterizing 462 enJSRVs insertion sites (including 435 newly described insertions) in the Ovis genus. We focused our study on endogenous copies inserted in the q13 locus of chromosome 6 (6q13). Those copies are known to confer resistance against exogenous JSRV thanks to alleles bearing a mutation in the gag gene. We characterized (i) the distribution of protective and non-protective alleles across Ovis species and (ii) the copy number variation of the 6q13 locus. Our results challenged the previous hypothesis of fixation and amplification of the protective copies in relation with domestication, and allowed building a new model for the evolution of the 6q13 locus. JSRV would have integrated the 6q13 locus after the Ovis-Capra divergence (5–11 MYA) and before the Ovis diversification (2.4–5 MYA). The protective mutation in the enJSRV 6q13 copy appeared shortly after its insertion and was followed by genomic amplifications, after the divergence between Pachyform lineage on one side and the Argaliform and moufloniform lineages on the other (2.4–5 MYA). Considering the potential selective advantage of the protective mutation, its fixation in both sheep and its closest wild relative Ovis orientalis may be due to natural selection before domestication from O. orientalis populations.
Similar content being viewed by others

Introduction
Retroviruses are viruses that integrate their genome into their host’s genome during their life cycle. This integration by horizontal transmission of the viral DNA may affect either somatic cells (exogenous retrovirus), or the germline leading then to a vertical transmission (endogenous retrovirus, ERV). Such endogenization is a quite common mechanism, since a large part of mammalian genomes is composed of retroviral sequences (8–10% (Mager and Stoye 2015)). These insertions play a major role in shaping genomes by altering gene structure, affecting their regulation or increasing genomic instability (Kaneko-Ishino and Ishino 2012). Some genes with an ERVs origin also play major physiological roles, especially during mammalian gestation, allowing fetal in vivo development (Lavialle et al. 2013). They are transmitted across generations in a mendelian way, and their loss, spreading or fixation are ruled by evolutionary forces. Thus, the evolutionary dynamics of neutral copies of viral DNA (e.g., no more functional due to mutations or transcriptionally repressed by epigenetic marks) is mainly driven by genetic drift, while insertions impacting host fitness are impacted by natural selection. Thus, studying neutral endogenous retroviruses copies allows to infer the demographic history of species (Sistiaga-Poveda and Jugo 2014) or populations (Chessa et al. 2009), whereas studying ERVs that alter the host fitness gives information about their adaptive histories.
The Jaagsiekte sheep retrovirus (JSRV) is a pathogenic agent responsible for the Ovine Pulmonary Adenocarcinoma, a transmissible lung cancer (Griffiths et al. 2010). Several endogenous JSRV copies (enJSRV) have already been described in the genome of several species of the subfamily Caprinae (Sistiaga-Poveda and Jugo 2014; Armezzani et al. 2014), demonstrating an old relationship between JSRV and small ruminants (at least 5–11 million years ago (MYA) (Hassanin et al. 2012)). The sheep, JSRV and enJSRV trio has long been used as a case study to address the complex relationships between retroviruses and their host, from functional and evolutionary perspectives (Sistiaga-Poveda and Jugo 2014). The sheep genome hosts several copies of enJSRV. Many of them remain uncharacterized, and twenty-seven have been described up to now (Armezzani et al. 2014). Among them, at least six have an insertion polymorphism in sheep populations (Chessa et al. 2009). EnJSRVs play a major role in sheep reproduction. The mRNA of the env enJSRV gene (coding for the envelope protein of the exogenous virus, which has a cell fusion inducing function) is required for the trophectoderm formation during gestation, and may play a role in the immunosuppression responsive of materno-fetal tolerance (Dunlap et al. 2006; Varela et al. 2009).
Besides this, two loci, enJSRV-20 and enJS56A1, interfere with the exogenous JSRV during host contamination. Both endogenous copies have alleles with an arginine (R) to tryptophan (W) mutation in the gag gene, encoding for a transdominant protein that interfere with the exogenous protein during the late step of viral replication (Arnaud et al. 2007). As a consequence, R/W gag mutant copies (R/W) have a protective effect against exogenous JSRV infection. These two copies are located in close proximity at the q13 locus on chromosome 6 (6q13) (Armezzani et al. 2011). The history of the 6q13 locus seems closely related to the evolution of the Ovis genus and two hypotheses emerge form previous works. According to the first hypothesis, two JSRV insertions would have occurred at 6q13 (Arnaud et al. 2007), after the Ovis-Capra divergence (i.e., 5–11 MYA, (Hassanin et al. 2012)), but before the divergence between the two main Ovis lineages separating the Pachyceriform group (i.e., O. nivicola Eschscholtz 1829, O. dali Nelson 1884 and O. Canadensis Shaw 1804) from the Moufloniform (O.vignei Blyth 1841, O. orientalis Gmelin 1774 and O. aries L. 1758) and Argaliform (O. ammon L. 1758) groups, 2.4–5 MYA (Rezaei et al. 2010). Under this hypothesis, the R/W mutation would have appeared in both enJSRVs copies (enJSRV-20 and enJS56A1) either independently or by gene conversion. Alternatively, the second hypothesis relies on the high sequence similarity between enJSRV-20 and enJS56A1, and present enJSRV-20 as the result of a recombination between enJS56A1 and another enJSRV, after an R/W gag mutation on enJS56A1 (Armezzani et al. 2014).
While enJS56A1 is described in all Ovis species, its protective allele is exclusive of the Moufloniform and Argaliform species and is described as fixed only in domestic animals (Arnaud et al. 2007). The distribution of enJSRV-20 across taxa is less clear. The insertion of enJSRV-20 is described as fixed in O. aries and O. orientalis, polymorph in O. ammon and O. canadensis and absent in O. dali and O. nivicola (no data available for O. vignei) (Armezzani et al. 2011). Moreover, it has a protective allele in O. aries, O. orientalis and O. ammon, which is fixed in O.aries and O.orientalis musimon Pallas 1762 (Arnaud et al. 2007; Armezzani et al. 2011).
The fixation of the protective alleles of both enJSRV-20 and enJS56A1 in domestic animals, has been hypothesized to be related to domestication, putatively due to promiscuity resulting from management in herds (Arnaud et al. 2007; Armezzani et al. 2011, 2014). Moreover, the 6q13 region carrying the protective enJSRV allele is duplicated several times, especially in domestic sheep (Armezzani et al. 2011). Because of the selective advantage of the R/W enJSRV allele, this increased number of copies could lead to an enhanced resistance via a dose effect. However, there is still no clear demonstration of the selective role of domestication on the fixation and duplication of the protective alleles. In this context, our study aimed at testing this hypothesis by assessing the link between the domestication status and the presence of protective alleles, taking advantage of the whole-genome sequencing of an unprecedented sampling of wild and domestic Ovis.
Materials and methods
Data sources
The references used were the genome assemblies of sheep (build Oar_v4.0 - GenBank assembly accession: GCA_000298735.2), goat (build Chir_2.0 - GenBank assembly accession: GCA_000317765.2), and JSRV (RefSeq assembly accession: GCF_000850005.1), as well as the 3′ and 5′ flanking sequences of enJS56A1 (F. Arnaud, personal communication, see supplementary information). The genomes of 76 individuals from five Ovis species were retrieved from the ENA archive (accession numbers in Supplementary table 1). Two O. dali, and three O. Canadensis individuals represented the Pachyform lineage while four O. vignei, eleven O. orientalis and forty-six O. aries represented the Argaliform and Moufloniform groups. Ovis orientalis individuals represented current Iranian populations from the domestication centre, while sheep were representative of several populations and breeds (Alberto et al. 2018, see Table 1).
Scan for enJSRV insertions
Reads from all animals were pooled and aligned on the JSRV reference genome with BWA mem algorithm (defaults parameters, Li and Durbin 2009, mapping profile on JSRV is presented in Supplementary Figure 1). Unmapped reads whose mate aligned with the highest mapping quality (60) on the JSRV genome were aligned on the sheep reference genome OAR-v4.0. We considered as insertion sites the regions of the sheep genome where reads aligned with high mapping quality (60) and a coverage exceeding 5×. To avoid multiple detections of the same insertion, regions distant by <10 kb were considered to be related to the same insertion. Based on these regions and the aligned reads, we built a presence/absence table for the 76 studied individuals. We only kept for further analyses the sites detected independently in at least two individuals.
Validation of the enJSRV insertion scan procedure
We applied several strategies to validate the enJSRV insertion scan procedure.
(i) We tested our ability to detect previously described polymorphic enJSRVs insertion site (Chessa et al. 2009). We identified insertion sites in the OAR-v3.1 reference genome by performing in silico amplifications with isPCR (Kuhn et al. 2012) (two mismatches authorized, 10 kb Max product size, primer sequences in supplementary table 2). We obtained corresponding sequences in OAR-v4.0 by alignment (BLASTn search, defaults parameters) (Camacho et al. 2009). After exploration of OAR-v3.1 for insertion sites of seven polymorphic enJSRVs (enJSRV-6, -7, -8, -15, -16, -18 and enJS5F16), we could localize five of them in OAR-v3.1 with a precise equivalent on OAR-V4.0 (i.e., enJSRV-6, -8, -16, -18 and enJS5F16 (supplementary table 2)). Thereafter, our insertion scan procedure detected all these enJSRVs but EnJSRV-8. This result is consistent with the distribution of this rare insertion found in Northern Europe (Chessa et al. 2009), a geographic area which was not covered by our sampling.
(ii) We map reads identified as enJSRVs reads on all known betaretroviruses present in Genbank, and found no match except with ENTV, close relative of JSRV, with no known endogenous copy in sheep (supplementary table 3).
(iii) We aligned JSRV genome against the reference Ovis genome (OAR_v4) using BLASTn software (Camacho et al. 2009). We identified 126 alignments longer than 100 bp. Clustering regions within 10 kb windows allowed identifying 68 enJSRVs insertions sites in the sheep reference genome. This order of magnitude is consistent with our estimation of 69–109 insertions per individual (see “global enJSRV survey” section below).
6q13 locus identification
We identified the 6q13 coordinates on the sheep and goat genomes by BLASTn alignments (with defaults parameters) of the 5′ and 3′ flanking enJSRV-20 sequences (372 and 342 bp long, respectively, F. Arnaud, personal communication), on the OAR-v4.0 and Chir_2.0 references, respectively.
Copy number estimation in the 6q13 region
The fragments constituting the library (Table 1) for paired-end sequencing were too short to allow mate-pairs to anchor both on the 6q13 region and on the enJSRV mutation site (see Supplementary Figure 2). Indeed, the mean fragment size ranged from 170 to 330 bp, while the R/W mutation was at 642 bp from the 5′ end of the JSRV sequence. Thus it was not possible to obtain direct evidence that a copy inserted in 6q13 region carried the protective mutation. We implemented an indirect strategy relying on the estimation for each individual of: (i) the number of inserted enJSRV copies at the 6q13 locus and (ii) the number of enJSRV copies carrying the R/W mutation. The insertion site at 6q13 was reconstructed by merging the 5′ and 3′ flanking sequences of enJSRV-20 with the JSRV sequence. we aligned reads from all individuals independently on this reconstructed sequence using the BWA mem algorithm (defaults parameters) (Li and Durbin 2009). Reads with a mapping quality lower than 60 were discarded.
(i) The number of inserted copies at the 6q13 locus (i.e., #enJSRV6q13) was estimated by counting read pairs with one read aligned on either the 5′ or the 3′ flanking sequence and its mate aligned on the JSRV sequence. This estimate was normalized to allow the comparison between individuals, and because of a possible bias due to the library construction or coverage. For this purpose, 100 contiguous pairs of 350 bp windows (i.e., the size of the 5′ and 3′ flanking sequences) were randomly selected in the genome. For each pair of windows, read pairs with one read correctly aligned (quality equal to 60) in each window were counted. The normalization consisted in dividing #enJSRV6q13 by the median number of reads correctly mapped on the 100 randomly selected pairs of windows.
(ii) For each individual, the number of copies carrying the R/W mutation was estimated by dividing the number of reads carrying the protective mutation (i.e., reads aligning perfectly to the JSRV genome but with a A at position 642 on the JSRV reference genome) by the mean whole genome coverage on OAR-v4.0.
Both estimations allowed assessing the correlation between the number of copies inserted in the 6q13 region and the number of copies carrying the mutation.
The number of contiguous 5′ end 3′ flanking sequences in the 6q13 region (i.e., showing no enJSRV insertion) was estimated by the number of pairs with one read aligned on the 5′ end of the sequence and its mate aligned on 3′ end, corrected with the same procedure as (i).
Analyses, statistical tests and graphs were made using R (Ihaka and Gentleman 1996).
Results
Global enJSRV survey
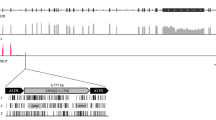
Among the 76 individuals from 5 Ovis species (including 56 O. aries), a total of 462 JSRV insertion sites distributed over all chromosomes (supplementary Figure 3, supplementary Table 4) were detected in at least two individuals (Fig. 1), with a median number of 90 insertions per individual (ranging from 69 to 109). Only 14 on the 462 insertions were found in all individuals of all species. There was a clear specific pattern of enJSRVs presence–absence (Fig. 1) with a significantly higher number of ERVs insertions in O. aries than in O. orientalis (medians of 93.5 and 85, respectively, t test, p-value: 0.048).

enJSRV presence (blue) absence (grey) heatmap. Individuals (lines) are ordered in species (Oa Ovis aries, Oo Ovis orientalis, Ov Ovis vignei, Oc Ovis canadensis, Od Ovis dalli) and enJSRV (columns) are ordered based on their relative binary distance. Only enJSRVs present in at least two individuals were kept. Layered barplot represent quantile 95 of coverage in insertion window. Red arrow indicates 6q13 insertion site
Study of the 6q13 locus
A BLASTn (Altschul et al. 1990) search for the sequences flanking the 5′ and 3′ ends of the enJS56A1 insertions allowed positioning the 6q13 region on the sheep and goat reference genomes. In goat, the insertion site was present three times on chromosome 6 (best hit between 6,129,894 and 6,130,613 bp) with no evidence for enJSRV insertion in any of the three duplications. In sheep, the 5′ and 3′ flanking sequences were present once in the contig UnplacedScaffold_004085138.1, between 10,443 and 10,819 bp, and 1 and 342 bp for the 3′ and 5′ flanking sequences, respectively. Interestingly, this insertion site was detected in all studied individuals with a coverage higher than that of all other insertion sites (quantile 95 of 25.56× when the median quantile 95 was 2.3× for all sites (Fig. 1)).
Because of the short size of the library inserts, we could not get direct evidence for the location of gag coding sequences (see supplementary Figure 2), we needed indirect information to infer the occurrence of R/W mutations at the 6q13 locus. We observed a strong correlation between the normalized number of inserted copies at 6q13 and the normalized number of putative enJSRV gag copies carrying the R/W mutation at the whole genome scale (Fig. 2a). Moreover, we found a week correlation between the number of enJSRV inserted at 6q13 and the number of enJSRV inserted elsewhere in the genome (Fig. 2b). Likewise, there were a week correlation between the proportions of reads supporting the R/W mutation and the number of enJSRV inserted across the whole genome (Fig. 2c). These findings strongly support the hypothesis that enJSRV copies carrying the R/W mutation are inserted within the 6q13 locus, even if we cannot exclude the occurrence of this mutation at a low frequency elsewhere in another enJSRV copy. Moreover, mate-pair data obtained for one sheep showed five paired reads with one read mapping on the sequence flanking the 3′ or 5′ side of the insertion and the other carrying the protective mutation. This confirmed the presence of the protective mutation at the 6q13 locus (Supplementary table 5).

Pairwise comparison of the number enJSRV-6q13, the total number of enJSRV in the genome and the number of R/W mutations. Relationship between the number of enJSRV in 6q13 region and the number of enJSRV R/W mutants in the genome (a), between the number of enJSRV in 6q13 region and the number of enJSRV in the genome (b) and between the number of enJSRV in the genome and the proportion of R/W sequences in the genome (c)
Within the 6q13 locus, the average number of enJSRV copies widely varied from 1 to 22 according to the species. North American species (O. Canadensis and O. dali) hosted between 1 and 4 (R) copies, while domestic sheep and Eurasian wild species (O. aries, O. orientalis and O. vignei) all had a highly variable number of protective (W) copies (i.e., between 2 and 30) (Fig. 2a). There were no significant difference between sheep (O. aries) and its closest wild relative (O. orientalis) for neither the number of insertions nor the number of protective copies. Conversely, in O. vignei, the number of insertions carrying R/W was lower than the number of inserted copies.
Because our data did not allowed differentiating enJSRV-20 and enJS56A1, we denote here both types of copies as enJSRV-6q13. The different Ovis species had different mean ratios between the number of protective copies and the number of enJSRV-6q13. For Ovis vignei, this ratio was 0.78, indicating a higher number of enJSRV-6q13 copies than protective mutations. O. orientalis and O. aries had ratios of 1.15 and 1.45, respectively, indicating a higher number of protective mutation than the number of enJSRV inserted within 6q13 region.
Interestingly, at the 6q13 locus the number of insertion sites without enJSRV was approximately twice the number of enJSRV in all species (Supplementary Figure 4). Despite the low number of American Ovis studied, we observed a clear difference between American and Eurasian species (Fig. 2a). American species (O. dali and O. canadensis) had a lower number of enJSRV-6q13 copies (harbouring or not the protective mutation) and a limited number of insertion sites without enJSRV, while Eurasian species (O. vignei, O. orientalis and O. aries) hosted a high number of mutated copies with an equivalent number of enJSRV inserted at 6q13 and twice more insertion sites without enJSRV.
Discussion
Methodological issues
The whole genome survey method described here is a reliable and simple strategy to detect mobile element insertions sites using paired-end data, inspired from the method developed by Keane et al. 2013. Nevertheless, this strategy encountered some limits, as the fragments constituting the library and the paired-end reads were too short to reconstruct the whole enJSRVs sequences. This limitation implies that we were not able to specify if each insertion corresponded to a full enJSRV, to a more or less degraded enJSRV sequence (as described in Arnaud et al. 2007), or to a solo LTR. Reconstructing the whole enJSRVs sequences was not the goal of our study, but would be mandatory to further characterize the impact of each insertion, or to date insertions by comparing mutations between 5′ and 3′ LTR sequences. This later question could be addressed using long read sequencing (with reads spanning several kb). However, information from our whole genome survey could be useful as a basis for future works on whole-genome characterization of enJSRV copies. Our method increased the number of enJSRVs insertion sites described from 27 (Arnaud et al. 2007) to 462. It also allowed overcoming limitations of other methods previously used. While the FISH method, traditionally used to detect and locate enJSRVs, only gives a rough insertion positioning at chromosomal scale (Carlson et al. 2003; Armezzani et al. 2011), whole genome surveys gave access to insertion site sequence with a much better resolution (100 bp compared to 10 kbp). Also, PCR based detections that were used to detect insertion polymorphism (Sistiaga-Poveda and Jugo 2014) are known to depend on the conservation of primer-biding sites among species, and may generate false polymorphism (e.g., false or null alleles (Pompanon et al. 2005)). In our approach, the length of the reads, the size of the fragments in the library and the sequencing depth increased the probability of alignment at insertion sites and consequently the detection power.
Endogenisation of JSRVs
Previous works already described a long-time interaction between the subfamily Caprinae and JSRV retroviruses, with enJSRVs shared by Ovis and Capra species (Arnaud et al. 2007; Sistiaga-Poveda and Jugo 2014). Among the 462 enJSRVs described in this work, 32 were present in all Ovis species among which 14 were present in all individuals. The fact that some enJSRVs insertions were present in all but a few individuals may reflect false-negative calls, which can be explained by heterogenous sequence coverages sometimes resulting in the absence of reads mapping on an insertion site in an individual. If this may be an issue for studying the fine scale distribution of enJSRV within individuals and populations, the current resolution allowed a clear characterization of species-specific patterns of enJSRVs insertions. Our results confirm that the first endogenisation of JSRV occured before the diversification of the Ovis genus more than 2.4 MYA (Rezaei et al. 2010). The occurrence of differential insertions among species highlights the fact that the endogenisation of JSRV accompanied the evolution of all Ovis species and is still active. The higher number of enJSRVs in sheep (O. aries) compared to wild species (and especially their closest wild relatives O. orientalis) may reflect the release of a purifying selection in domestic, and/or an increased exposure to exogenous JSRV with domestication related to the proximity between animals as the result of management in large herds.
History of the 6q13 region
The 6q13 region and its enJSRVs copies (i.e., enJSRV-20 and enJS56A1 here named enJSRV-6q13) played a special role in the evolution of the JSRV and sheep relationships. A protective mutation affecting these copies encodes for a transdominant protein that interfere with the exogenous protein during the late replication step, and is responsible for a genetic resistance against the exogenous JSRV (Armezzani et al. 2014). Our results allowed retracing the evolutionary history of this region.
We located the 6q13 region in an unplaced contig (i.e., UnplacedScaffold_004085138.1) of the sheep genome (Oar_v4.0), and showed that all Ovis individuals presented at least one endogenous JSRV in this region. In goat, we found no evidence for provirus insertion at this locus, as the 5′ and 3′ flanking sequences of enJS56A1 were contiguous. Moreover, these insertion sites were part of a 15 kb region repeated in tandem at least three times. In Ovis, the number of insertion sites without enJSRV was approximately twice the number of enJSRV in all species (Supplementary Figure 4). Thus, this triplication observed in sheep goats would have occurred before the Ovis-Capra divergence (5–11 MYA).
Taken together, these results confirmed that the insertion of JSRV in the 6q13 region occurred after the Ovis-Capra divergence (5–11 MYA) but before the diversification of the Ovis genus (2.4 MYA) (Rezaei et al. 2010). We detected the protective arginine to tryptophan (R/W) mutation previously reported in enJSRV-6q13 (Armezzani et al. 2014) in all individuals from the Eurasian species we analysed (O. aries, O. oritenalis and O. vignei). It was also detected in an O. canadensis. Even if we cannot exclude a sequencing error, as this observation is only supported by one single read, the hypothesis of a rare tryptophan mutant in the Bighorn sheep is likely, given the low number of studied individuals. In any case, the wide spread of the protective allele in all Eurasian species and its possible presence in the American species date the protective mutation during or soon after the separation between those two lineages (Rezaei et al. 2010). Studying more genomes from Ovis ammon, Ovis nivicola and the American species would allow dating this mutation more precisely.
Interestingly, we detected a genomic amplification within the 6q13 region, and found that the number of enJSRV-6q13 was correlated to the number of protective mutations (Fig. 2a). The most parsimonious scenario explaining those observations is the occurrence of the R/W mutation in the common ancestor of all Eurasian species, followed by genomic amplifications of the 6q13 region, between 1.26 and 2.42 MYA (Rezaei et al. 2010).
The distribution of the number of the protective allele per indivdual was similar between sheep (O. aries) and their closest wild relatives (O. orientalis), but was lower in O. vignei (Fig. 2a). This highlight the fact that contrary to O. orientalis and O. aries, some enJSRV-6q13 in O. vignei did not carry the protective mutation. This indicates a fixation of the protective copy in O. orientalis after the divergence with O. vignei (1.26 MYA) but before sheep domestication (10 KYA). Besides, the ratio between the number of protective mutations and the number of enJSRV-6q13 was higher than 1 in O. orientalis and O. aries. This suggests the occurrence of more R/W copies than enJSRV-6q13. This could be explained by considering that enJSRV-20 (which exact sequence remains uncharacterized) would be a recombination between enJS56A1 and an other enJSRV (Armezzani et al. 2014), allowing two protective mutations per enJSRV-20 insertion. A strong selection for an increase of the number of protective copies after the separation between O. vignei and the other Eurasian species would induce both the fixation of the protective allele and the spread of enJSRV-20 in the O. orientalis lineage.
A previous publication suggest that the protective allele (carrying the R/W mutation) was fixed in the period surrounding domestication (Arnaud et al. 2007). A more recent work hypothesized that the genomic amplification impacting the 6q13 region was specific to domestic sheep (Armezzani et al. 2011), and that the fixation of the protective allele was a consequence of selection related to the domestication process (Armezzani et al. 2011, 2014). In our study, sheep (Ovis aries) and their closest wild relatives (Ovis orientalis) host a similar number of protective enJSRV-6q13, which greatly varies among individuals. There was no distinction between wild and domestic animals, challenging the hypothesis of an impact of domestication on this locus. Indeed, even if we cannot state if amplified enJSRV-6q13 are copies of enJS56A1, enJSRV-20 or any other provirus sharing the same 3′ sequence, the most parsimonious scenario implies that the polymorphism observed in domestics reflects that of the wild group, and was captured during domestication about 10 KYA.
Caprinae and JSRV are engaged in a long-term relationship. As demonstrated by old and shared enJSRVs insertions sites among Ovis, this relationship was engaged before the Caprinae speciation (5–11 MYA). One of these old enJSRV integrated the 6q13 locus in Ovis genomes after the Ovis-Capra divergence (5–11 MYA, Rezaei et al. (2010)) and before the Ovis diversification (2.4–5 MYA, Rezaei et al. (2010)). This relation took a new turn when the protective mutation, conferring resistance against exogenous JSRV occurred shortly after its insertion in Ovis on the 6q13-inserted enJSRVs. This mutation was followed by genomic amplifications, after the divergence between Pachyform lineage on one side and the Argaliform and moufloniform lineages on the other (2.4–5 MYA, Rezaei et al. (2010)). Considering the selective advantage of the protective mutation, natural selection may account for the widespread amplification of this locus in all Moufloniform and Argaliform species. Genomic amplification is well known as being a mechanism involved in various types of resistance, for example against nematode in cattle (Hou et al. 2012) or chemical insecticides in mosquitoes (Faucon et al. 2015). If resistance against JSRV infection rely on a dose-effect of enJSRV-6q13 env expression (Viginier et al. 2012), then an increase in the number of copies would increase individual fitness and would be selected. Finally, the recent discovery of enJSRV-26, which is able to escape enJSRV-6q13 resistance in the Texel breed, (Armezzani et al. 2011) and may be a step of the “arm race” between sheep and JSRV.
Data archiving
Sequences used in this work are available at: http://projects.ensembl.org/nextgen.
References
Alberto FJ, Boyer F, Orozco-terWengel P, Streeter I, Servin B, Villemereuil P et al. (2018) Convergent genomic signatures of domestication in sheep and goats. Nat Commun 9:813
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Armezzani A, Arnaud F, Caporale M, di Meo G, Iannuzzi L, Murgia C et al. (2011) The signal peptide of a recently integrated endogenous sheep betaretrovirus envelope plays a major role in eluding Gag-Mediated Late Restriction. J Virol 85:7118–7128
Armezzani A, Varela M, Spencer TE, Palmarini M, Arnaud F (2014) “Ménage à Trois”: the evolutionary interplay between JSRV, enJSRVs and domestic sheep. Viruses 6:4926–4945
Arnaud F, Caporale M, Varela M, Biek R, Chessa B, Alberti A et al. (2007) A paradigm for virus–host coevolution: sequential counter-adaptations between endogenous and exogenous retroviruses. PLoS Pathog 3:e170
Arnaud F, Murcia PR, Palmarini M (2007) Mechanisms of late restriction induced by an endogenous retrovirus. J Virol 81:11441–11451
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al. (2009) BLAST+: architecture and applications. BMC Bioinform 10:421
Carlson J,Lyon M,Bishop J,Vaiman A,Cribiu E,Mornex J.-F, et al(2003) Chromosomal distribution of endogenous Jaagsiekte sheep retrovirus proviral sequences in the sheep genome J Virol 77:9662–9668
Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I et al. (2009) Revealing the history of sheep domestication using retrovirus integrations. Science 324:532–536
Dunlap KA, Palmarini M, Varela M, Burghardt RC, Hayashi K, Farmer JL et al. (2006) Endogenous retroviruses regulate periimplantation placental growth and differentiation. Proc Natl Acad Sci USA 103:14390–14395
Faucon F, Dusfour I, Gaude T, Navratil V, Boyer F, Chandre F et al. (2015) Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res 25:1347–1359
Griffiths DJ, Martineau HM, Cousens C (2010) Pathology and pathogenesis of ovine pulmonary adenocarcinoma. J Comp Pathol 142:260–283
Hassanin A, Delsuc F, Ropiquet A, Hammer C, Jansen van Vuuren B, Matthee C et al. (2012) Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. C R Biol 335:32–50
Hou Y, Liu GE, Bickhart DM, Matukumalli LK, Li C, Song J et al. (2012) Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct Integr Genom 12:81–92
Ihaka R, Gentleman R (1996) R: a language for data analysis and graphics. J Comput Graph Stat 5:299–314
Kaneko-Ishino T, Ishino F (2012). The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol 3:262.
Kuhn RM, Haussler D, Kent WJ (2013). The UCSC genome browser and associated tools. Brief Bioinform 14: 144–161.
Lavialle C, Cornelis G, Dupressoir A, Esnault C, Heidmann O, Vernochet C et al. (2013) Paleovirology of ‘syncytins’, retroviral env genes exapted for a role in placentation. Philos Trans R Soc Lond B Biol Sci 368:20120507
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Mager DL, Stoye JP (2015). Mammalian endogenous retroviruses. Microbiol Spectr 3:1–20
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6:847–846
Rezaei HR, Naderi S, Chintauan-Marquier IC, Taberlet P, Virk AT, Naghash HR et al. (2010) Evolution and taxonomy of the wild species of the genus Ovis (Mammalia, Artiodactyla, Bovidae). Mol Phylogenet Evol 54:315–326
Sistiaga-Poveda M, Jugo BM (2014) Evolutionary dynamics of endogenous Jaagsiekte sheep retroviruses proliferation in the domestic sheep, mouflon and Pyrenean chamois. Heredity 112:571–578
Varela M, Spencer TE, Palmarini M, Arnaud F (2009) Friendly viruses: the special relationship between endogenous retroviruses and their host. Ann N Y Acad Sci 1178:157–172
Viginier B, Dolmazon C, Lantier I, Lantier F, Archer F, Leroux C et al. (2012) Copy number variation and differential expression of a protective endogenous retrovirus in sheep (J-P Vartanian, Ed.). PLoS ONE 7:e41965
Keane TM, Wong K, Adams DJ (2013). RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics 29:389–390
Acknowledgements
We thank Frederic Arnaud for providing us enJSRV-20 flanking sequences and useful suggestions. We also thank Christophe Terzian for the help provided throughout the study. This work was supported by the European Union 7th framework project NEXTGEN (Grant Agreement no. 244356). LECA is part of the Labex OSUG@2020 (ANR 10LABX56). Most of the computations presented in this paper were performed using the CIMENT infrastructure (https://ciment.ujf-grenoble.fr), which is supported by the Rhône-Alpes region (GRANT CPER07_13 CIRA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Cumer, T., Pompanon, F. & Boyer, F. Old origin of a protective endogenous retrovirus (enJSRV) in the Ovis genus. Heredity 122, 187–194 (2019). https://doi.org/10.1038/s41437-018-0112-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0112-z
