
Abstract
Purpose
Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of chronic kidney disease in childhood and adolescence. We aim to identify novel monogenic causes of CAKUT.
Methods
Exome sequencing was performed in 550 CAKUT-affected families.
Results
We discovered seven FOXC1 heterozygous likely pathogenic variants within eight CAKUT families. These variants are either never reported, or present in <5 alleles in the gnomAD database with ~141,456 controls. FOXC1 is a causal gene for Axenfeld–Rieger syndrome type 3 and anterior segment dysgenesis 3. Pathogenic variants in FOXC1 have not been detected in patients with CAKUT yet. Interestingly, mouse models for Foxc1 show severe CAKUT phenotypes with incomplete penetrance and variable expressivity. The FOXC1 variants are enriched in the CAKUT cohort compared with the control. Genotype–phenotype correlations showed that Axenfeld–Rieger syndrome or anterior segment dysgenesis can be caused by both truncating and missense pathogenic variants, and the missense variants are located at the forkhead domain. In contrast, for CAKUT, there is no truncating pathogenic variant, and all variants except one are located outside the forkhead domain.
Conclusion
We thereby expanded the phenotype of FOXC1 pathogenic variants toward involvement of CAKUT, which can potentially be explained by allelism.
Similar content being viewed by others

INTRODUCTION
Congenital anomalies of the kidneys and urinary tract (CAKUT) are the most common cause of chronic kidney disease (CKD) in the first three decades of life.1 CAKUT can present as an isolated renal or genitourinary condition or as part of a clinical syndrome.2,3 To date, 40 genes have been identified as monogenic causes of isolated CAKUT (Supplementary Table S1), and 153 genes have been identified for syndromic CAKUT (Supplementary Table S2).4,5
FOXC1 belongs to the group of FOX proteins featuring a forkhead domain, which is a winged helix protein domain consisting of 80 to 100 amino acids.6 The forkhead domain is known to bind DNA and through this binding regulate DNA transcription.7,8 Heterozygous FOXC1 pathogenic variants (Fig. 1a) have been identified as the cause of Axenfeld–Rieger syndrome type 3 (OMIM 602482) and anterior segment dysgenesis 3 (OMIM 601631). The gene was first found to cause Axenfeld–Rieger anomaly,9,10,11 which is an anomaly with variable and overlapping presentations of posterior embryotoxon, peripheral iris attachments to Schwalbe’s line and the cornea, and iris abnormalities.12,13 Later studies indicated that FOXC1 pathogenic variants may cause other anterior segment anomalies that are not Axenfeld–Rieger anomaly.10,13,14,15,16 These variable anterior segment anomalies include aniridia, iris hypoplasia, primary congenital glaucoma, corneal and lens abnormalities, and are collectively called anterior segment dysgenesis (Fig. 1a).10,13,14,15,16 Syndromic cases may present with brain abnormalities, cardiac and vascular malformations, dysmorphic features, dental anomalies, sensorineural hearing loss, and orthopedic abnormalities.17,18,19,20

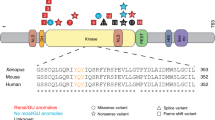
There are truncating and missense pathogenic variants in anterior segment dysgenesis, and all missense pathogenic variants except one are in the forkhead (FH) domain. In contrast, in individuals with CAKUT we detected no truncating pathogenic variants, and all pathogenic variants except one are located out of the FH domain. (a) FOXC1 pathogenic variants and phenotypes published in the literature for anterior segment dysgenesis. A1. Blue horizontal boxes and lines represent pathogenic truncating variants caused by out-of-frame indels. The junction of each box and line corresponds to the start of amino acid change caused by frame shift. The right end of each line corresponds to the predicted termination position of the protein. A2. Black horizontal lines represent the extent of pathogenic truncating variants caused by nonsense pathogenic variants. The right end of each line corresponds to the predicted termination position of the protein. A3. Green vertical lines represent pathogenic in-frame indels. The location of each vertical line corresponds to the position of the in-frame change. A4. Red vertical lines represent pathogenic substitution variants caused by missense pathogenic variants. The location of each vertical line corresponds to the position of the missense pathogenic variant. All but one missense pathogenic variant was located within the FH domain. In contrast, nonsense pathogenic variants, in-frame and out-of-frame indels are not limited in FH domain. (b) FOXC1 pathogenic variants and phenotypes in CAKUT cohort. There is a spectrum of CAKUT phenotypes that is described in detail for each family. B1. Each red box represents a CAKUT family with a pathogenic missense variant. Each red arrow points to the position of the substitution. The individual ID, CAKUT and extrarenal phenotypes, nucleotide and amino acid change are presented in each box. B2. Each green box represents a CAKUT family with a pathogenic in-frame indel. Each green arrow points to the position of the indel. The individual ID, CAKUT and extrarenal phenotypes, nucleotide and amino acid change are presented in each box. There is no nonsense pathogenic variant or out-of-frame indels found in the CAKUT cohort. All pathogenic variants except one in-frame deletion are located out of FH domain. B3. Patients presented with syndromic CAKUT have FOXC1 pathogenic variants located between the start site to the end of the FH domain. Patients presented with isolated CAKUT have FOXC1 pathogenic variants downstream to the FH domain. (c) Transcript and protein domain diagram of FOXC1. C1. FOXC1 transcript NM_001453. There is no 5’ UTR and there is only one exon. C2. FOXC1_Human protein UniProtKB–Q12948. The yellow octagon represents the FH domain. Pink rectangles represent low complexity detected by the SEG program (Wootton JC and Federhen S.41). AVM arteriovenous malformation, Bil bilateral, CKD chronic kidney disease, DD developmental delay, ESRD end-stage renal disease, FTT failure to thrive, LKS left kidney stone, Lt left, MCDK multicystic dysplastic kidney, n/d no data, PUV posterior urethral valve, Rt right, UPJO ureteropelvic junction obstruction, VUR vesicoureteral reflux.
So far, CAKUT have not been associated with FOXC1. However, mouse models for FOXC1 show severe CAKUT phenotypes with extrarenal malformations.21,22,23,24,25 CAKUT phenotypes in these mouse models exhibit variable expressivity, and the mode of inheritance (MOI) is autosomal dominant with incomplete penetrance.21,22,23,24,25
Employing exome sequencing (ES) analyses, we identified heterozygous FOXC1 likely pathogenic variants in eight families with CAKUT, which were absent from a control cohort with steroid-resistant nephrotic syndrome (SRNS). We thereby expand the phenotype of heterozygous FOXC1 pathogenic variants to include CAKUT. We showed evidence that involvement with CAKUT happens on an allelic basis, where in Axenfeld–Rieger syndrome or anterior segment dysgenesis, phenotypes can be caused by both FOXC1 truncating and missense variants, and the missense variants are located at the forkhead domain; whereas in contrast, for patients with CAKUT, there is no FOXC1 truncating variant, and all pathogenic variants except one are located out of the forkhead domain.
MATERIALS AND METHODS
Study participants
The study was approved by the institutional review board of Boston Children’s Hospital as well as the institutional review boards of institutions where we have recruited families. The study has been performed in accordance with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. From January 2010 to January 2019, patients with CAKUT were enrolled after obtaining informed consent. Following informed consent, 550 different families were enrolled and had ES performed on DNA samples. All patients with CAKUT were referred to us by their pediatric nephrologist or urologist who made a clinical diagnosis of CAKUT on the basis of renal imaging studies. CAKUT was defined as demonstration of any abnormality of number, size, shape, or anatomic position of the kidneys, gonads, or other parts of the urinary tract that included at least one of the following: renal agenesis, renal hypo-/dysplasia, multicystic dysplastic kidneys, hydronephrosis, ureteropelvic junction obstruction, hydroureter, vesicoureteral reflux (VUR), ectopic or horseshoe kidney, duplex collecting system, ureterovesical junction obstruction, epi-/hypospadias, posterior urethral valves (PUV), and cryptorchidism.
Control cohort
The control cohort consisted of 100 families with SRNS due to an underlying monogenic cause (Supplementary Table S3).4 The participants in the cohort were carefully evaluated to ensure that there were no CAKUT phenotypes present.
Exome sequencing (ES) and variant calling
ES was performed as previously described.26 In brief, genomic DNA was isolated from blood lymphocytes or saliva samples and subjected to exome capture using Agilent SureSelect human exome capture arrays (Life Technologies) followed by next-generation sequencing on the Illumina HighSeq sequencing platform. Sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19), and variants were called using CLC Genomics Workbench (version 6.5.2) software (CLC Bio, Aarhus, Denmark).
Variant filtering
Variants were filtered for most likely deleterious variants, as previously described.27,28 Variants with minor allele frequencies >1% in the dbSNP (version 147) or the 1000 Genomes Project (2504 subjects of various ethnicities; October 2015 data release) databases were excluded because they were unlikely to be deleterious. Synonymous and intronic variants that were not located within splice site regions were excluded. Retained variants, which included nonsynonymous variants and splice site variants, were then analyzed.
Screening for genes known to cause CAKUT
We evaluated ES data for causative pathogenic variants in 40 monogenic genes for isolated CAKUT, and in 153 monogenic genes for syndromic CAKUT known at the time (Supplementary Tables S1 and S2).4
Variant evaluation to identify novel monogenic causes of CAKUT
Modified from our previous publication,4 the remaining variants were evaluated to meet the following criteria to be determined pathogenic:
Criterion 1
Evolutionary conservation or functional severity prediction: The variant needs to have a CADD score >20, OR be predicted to be deleterious by two of three in silico prediction tools, OR have a moderate to strong evolutionary amino acid conservation at least to Xenopus tropicalis.
In criterion 1, CADD scores are computed based on the latest algorithm published by Rentzsch et al.29 The three in silico prediction tools we incorporated for evaluation are MutationTaster,30 PolyPhen-2,31 and SIFT.32 Orthologous proteins across phylogeny used to evaluate evolutionary conservation were obtained from the Ensembl Genome Browser and were aligned using the Clustal Omega multiple sequence alignment tool (EMBL-EBI).33
Criterion 2
Allele frequency: The variant needs to present in fewer than 20 heterozygous alleles in the general population database gnomAD.
In criterion 2, variants were evaluated based on the population frequency using gnomAD, which is a population genome database consisting of 141,456 control individuals.34 Allele frequency of less than 20 heterozygous alleles in gnomAD is an arbitrary cut-off we have been applying to autosomal dominant diseases like CAKUT that have a strong component of incomplete penetrance and variable expressivity.4
Implementation of structural data for variant evaluation
Protein domain structure cartoons and evaluation were based on the UniProt database.35 The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Statistical analysis
Statistical analysis was performed with STATA (College Station, TX). Two-sample categorical variables were compared using two-tailed Fisher’s exact test. One-sample categorical variables were compared with the theoretical null proportion by one-sample proportion test. A P value < 0.05 was considered statistically significant.
Web resources
CADD (Combined Annotation Dependent Depletion) https://cadd.gs.washington.edu/
Clustal Omega http://www.ebi.ac.uk/Tools/msa/clustal
Ensembl Genome Browser http://www.ensembl.org
Exome Variant Server http://evs.gs.washington.edu/EVS
Genome Aggregation Database (gnomAD) http://gnomad.broadinstitute.org
HGMD Professional 2016.3 https://portal.biobase-international.com/hgmd
Homozygosity Mapper http://www.homozygositymapper.org/
MutationTaster http://www.mutationtaster.org
Online Mendelian Inheritance in Man (OMIM) http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2
Sorting Intolerant From Tolerant (SIFT) http://sift.jcvi.org
UCSC Genome Browser http://genome.ucsc.edu/cgi-bin/hgGateway
Uniprot Consortium http://www.uniprot.org/
1000 Genomes Browser http://browser.1000genomes.org
RESULTS
FOXC1 pathogenic variants identified in patients with CAKUT
By ES analyses, we discovered seven FOXC1 pathogenic variants in eight CAKUT families (Table 1 and Fig. 1). All individuals carrying the FOXC1 pathogenic variants are heterozygote. None of the seven pathogenic variants were reported before in patients with Axenfeld–Rieger syndrome, anterior segment dysgenesis, or congenital glaucoma. Two of the seven pathogenic variants are novel, i.e., they were never observed in the population database before, including the gnomAD database that collects 141,456 control individuals.34 The other five pathogenic variants, though reported in the population database, are present in less than five individuals as a heterozygote.
Among the seven pathogenic variants found in the CAKUT patients, three are in-frame indels, and the other four are missense variants (Table 1, Fig. 1). Five of seven have a CADD score greater than 20, indicating they are predicted to be among the top 1% most deleterious variants in the human genome. One of the other two pathogenic variants, c.1090C>T, p.Pro364Ser, has a CADD score close to 20 (19.23), which is predicted to be among the top 1.19% most deleterious variants. This variant is conserved to zebrafish (Danio rerio). The other pathogenic variant, c.1139G>T, p.Gly380Val, has a lower CADD score at 16.45, which is predicted to be among the top 2.2% most deleterious. It is predicted to be deleterious/disease-causing by both SIFT and MutationTaster, and has never been observed in the population database before.
All eight families were then screened for genes known to cause either nonsyndromic isolated CAKUT or syndromic CAKUT (Supplementary Tables S1 and S2). No likely causative heterozygous or biallelic variants were detected in those genes.
Phenotype spectrum in patients carrying FOXC1 pathogenic variants
Families identified with FOXC1 pathogenic variants in our cohort demonstrated a wide spectrum of CAKUT phenotypes (Table 1 and Fig. 1). The CAKUT phenotypes include unilateral renal agenesis, bilateral renal dysplasia, unilateral multicystic dysplastic kidney, ureteropelvic junction obstruction, hydronephrosis, VUR, and PUV (Table 1). Among these presentations, VUR is the most common one, as seen in three of eight families, and PUVs were seen in two of eight families. Of notice, one individual (A5071–31) has left hydronephrosis first found in prenatal ultrasound, and serial follow-ups showed the hydronephrosis resolved at the age of 2 years (Table 1).
Regarding extrarenal manifestations, five of eight families have isolated CAKUT with no extrarenal features, while the other three families have syndromic CAKUT. These include anomalies in eyes, blood vessels, brain, bones, or facial dysmorphologies (Fig. 1b, Table 1). Of note, pathogenic FOXC1 variants of families with syndromic CAKUT (A3938, A3859, B1252) are located at or proximal to the forkhead domain (Fig. 1b), while the pathogenic variants of the families with isolated CAKUT are located distal to the forkhead domain (Fig. 1b). Even though this distribution of the variants for syndromic versus isolated CAKUT is statistically significant (z = 2.000, P = 0.0228), the numbers are too small to draw a meaningful conclusion.
There are two families present with eye features (A3859, B1252) who would potentially fulfill the diagnostic criteria of Axenfeld–Rieger syndrome/anterior segment dysgenesis. However, we were not able to follow up these patients to examine if they have anterior segment dysgenesis or not. In three families (A1252, B1402, and A5071), the pathogenic variants are inherited from an unaffected parent. This observation suggests incomplete penetrance and variable expressivity, which has been well documented in CAKUT,36,37 and has been repeatedly observed in FOXC1 mouse model.21,22,23,24,25
FOXC1 pathogenic variants are absent from a control cohort
To test for the significance of the ES finding, we screened for FOXC1 variants in a negative control cohort of 100 families who had ES performed for SRNS, and who were identified to have a causative pathogenic variant in one of the 55 established nephrotic syndrome (NS) genes (Supplemental Table S3).38 The individuals in this negative control cohort have been carefully phenotyped to exclude CAKUT.
The same evaluation criteria were applied to FOXC1 variants in this negative control cohort as in the CAKUT case cohort (see “Materials and methods”). Following the first filtering by 1% allele frequency, only one variant remained. Using Fisher’s exact test, the odds ratio was calculated as 8.185, with a P = 0.0083 (Table 2). This confirmed that FOXC1 variants are strongly enriched in the CAKUT cohort. When further evaluated to determine if the variant is pathogenic (using the same evaluation criteria as in the CAKUT case cohort, see “Variant evaluation to identify novel monogenic causes of CAKUT” and “Materials and methods”), no variant from the SRNS control cohort could be retained. In conclusion, pathogenic variants in FOXC1 are absent from the negative control cohort (Table 2).
Genotype–phenotype correlation
None of the seven pathogenic variants that we identified in the CAKUT cohort were ever reported previously in patients with Axenfeld–Rieger syndrome or anterior segment dysgenesis (Fig. 1a; detailed data available from authors upon request). To evaluate whether the presence of CAKUT phenotype in FOXC1 pathogenic variants is due to allelism, we conducted genotype–phenotype correlations as follows.
Excluding cytogenic or gross chromosomal deletion/insertion/duplication/translocation that encompasses FOXC1, there are 77 pathogenic FOXC1 variants reported to cause Axenfeld–Rieger syndrome or anterior segment dysgenesis (Fig. 1a). The class of pathogenic variants was as follows (Fig. 1a): truncating variants by out-of-frame indels (Fig. 1A1, blue), truncating variants by nonsense single-nucleotide variant (SNV) (Fig. 1A2, black), substitution variants by in-frame indels (Fig. 1A3, green), and substitution variants by missense SNV (Fig. 1A4, red).
Figure 1A4 demonstrates that all but one pathogenic FOXC1 missense variants causing Axenfeld–Rieger syndrome or anterior segment dysgenesis are located in the forkhead domain (Fig. 1A4). On the other hand, the truncating variants, either caused by nonsense SNVs or out-of-frame indels, stop at different locations along the transcript (Fig. 1A1, Fig. 1A2).
In contrast, in our CAKUT cohort, we did not observe any truncating pathogenic variant, i.e., no nonsense SNVs, and no frame-shift indels. The class of pathogenic variants was as follows (Fig. 1b): substitution variants by missense SNV (Fig. 1B1, red), and substitution variants by in-frame indels (Fig. 1B2, green). The locations of these pathogenic variants do not cluster in the forkhead domain. In fact, all pathogenic variants except one are located out of the forkhead domain (Fig. 1b).
DISCUSSION
By ES, we identified heterozygous FOXC1 likely pathogenic variants in eight families with CAKUT compared with the control cohort with no CAKUT phenotype. We found that FOXC1 rare variants (<1% minor allele frequency) are overrepresented among patients with CAKUT, and the likely pathogenic variants are absent in the control cohort. We further showed evidence that involvement with CAKUT happens on an allelic basis, where in Axenfeld–Rieger syndrome or anterior segment dysgenesis, phenotypes can be caused by both truncating and missense variants, and the missense variants are located at the forkhead domain; whereas in contrast, for patients with CAKUT, there is no truncating variant, and all pathogenic variants except one are located out of the forkhead domain.
Mouse models for FOXC1
Grüneberg reported the first mouse model for Foxc1.39 By examining mice deceased at birth, he found that the mutant mice presented with hydrocephalus with massively enlarged and hemorrhagic cerebral hemispheres. The gene demonstrates pleiotropism with cartilage and bone phenotypes. By counting the death at birth, he categorized this pathogenic variant as autosomal recessive. Green evaluated the genotypes and examined the viscera of the mice.21 For homozygotes, they recapitulated Grüneberg’s findings that these mice die at birth, and first found these mice had severe urogenital abnormalities. In heterozygotes, the mice presented with incomplete penetrance and variable expressivity with a wide spectrum of CAKUT.21
In 1998, Kume et al. first expanded the phenotype of these Foxc1 mice to include eye malformations.22 They thereby confirmed Foxc1 to be the causal gene of Axenfeld–Rieger syndrome type 3 (OMIM 602482) and anterior segment dysgenesis 3 (OMIM 601631) in the mouse model.22 For other organ systems, they reported identical phenotypes as Green,21 including the findings in the genitourinary system in mice.22
In 2000, Kume et al. further investigated the pathogenic Foxc1 variants in mice with different genetic backgrounds.23 They demonstrated variable expressivity and incomplete penetrance of CAKUT again, but the penetrance varied across different strains of the mice.23 This incomplete penetrance under different genetic backgrounds was reproduced by Komaki et al.24 and Motojima et al.25 using different inbred mice strains.
Incomplete penetrance, variable expressivity, and transient developmental phenotype in FOXC1 pathogenic variants
Similar to the findings in the mouse models, we here found evidence of incomplete penetrance and variable expressivity in our human CAKUT cohort (Fig. 1 and Fig. 2). Families with FOXC1 pathogenic variants in our cohort demonstrated a spectrum of CAKUT phenotypes, ranging from renal agenesis, renal dysplasia, multicystic dysplastic kidney, ureteropelvic junction obstruction, hydronephrosis, VUR, to PUV (Table 1 and Fig. 1). These demonstrated a variable expressivity.

The families are sorted based on the position of their pathogenic variants. Amino acid change is included in each pedigree. CAKUT phenotypes are labeled in green. Extrarenal phenotypes are labeled in blue. AVM arteriovenous malformation, CAKUT congenital anomalies of kidney and urinary tract, CKD chronic kidney disease, ESRD end-stage renal disease, FTT failure to thrive, MCDK multicystic dysplastic kidney, n/d no data, PUV posterior urethral valve, UPJO ureteropelvic junction obstruction, VUR vesicoureteral reflux.
In family B1252, FOXC1 c.433_435del segregated from the mother to the proband. However, the mother presented with only high myopia, skeletal deformity, and cerebral arteriovenous malformations (AVM) but no CAKUT phenotype. This observation suggests incomplete penetrance. Incomplete penetrance was also found in the proband B1402–21 and the father B1402–11, and in the proband A5071–31 and the mother A5071–22. (Fig. 1 and Fig. 2)
Of note, in A5071–31 the CAKUT phenotype of left hydronephrosis was discovered by prenatal ultrasound. The infant was followed up closely after birth, and the hydronephrosis resolved at the age of two years. In this case, CAKUT presented as a transient developmental phenotype and resolved as the infant grew. It is hard to trace back to see if the mother with the same genotype (A5071–22) had similar kidney developmental problems prenatally. Incomplete penetrance and variable expressivity are a well-known genetic phenomenon in CAKUT.36,37 The discrepancy that deletions of FOXC1 in mice resulted in CAKUT, whereas we detected no deletions in humans with CAKUT, can only be assessed once more pathogenic variants in patients with CAKUT have been discovered.
FOXC1 and CAKUT in human: potential risk allele for CAKUT
The association of FOXC1 variants to CAKUT in humans has been described once previously.40 Nakano et al. identified an insertion of a triplet GGC in FOXC1 in three of the seven patients with CAKUT.40 We reviewed their Sanger sequencing trace and found the variant they found is FOXC1 c.1139_1141dup, which causes an in-frame duplication of Glycine (p.Gly380dup). However, with the population database available,34 we know this variant has an allele frequency of 15.37% in the general population. Therefore, this variant would not establish a Mendelian monogenic cause of human CAKUT with certainty. In our study, we demonstrated that FOXC1 rare variants are overrepresented in the CAKUT cohort compared with the control cohort (odds ratio [OR] 8.185, P = 0.0083) (Table 2).
Conclusion
We propose a phenotype expansion of FOXC1 to include CAKUT. On the basis that heterozygous pathogenic variants in the CAKUT cohort are distinct from the variants in Axenfeld–Rieger syndrome or anterior segment dysgenesis, our finding is most likely explained by allelism. Axenfeld–Rieger syndrome or anterior segment dysgenesis can be caused by both truncating and missense pathogenic variants, and the missense pathogenic variants are located at the forkhead domain. In contrast in CAKUT, there is no truncating pathogenic variant, and all pathogenic variants except one are located out of the forkhead domain. We hereby expand the phenotype of pathogenic FOXC1 variants to also include CAKUT.
References
Chesnaye N, Bonthuis M, Schaefer F, et al. Demographics of paediatric renal replacement therapy in Europe: a report of the ESPN/ERA–EDTA registry. Pediatr Nephrol. 2014;29:2403–2410.
Vivante A, Kohl S, Hwang D-Y, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014;29:695–704.
Soliman NA, Ali RI, Ghobrial EE, Habib EI, Ziada AM. Pattern of clinical presentation of congenital anomalies of the kidney and urinary tract among infants and children. Nephrology. 2015;20:413–418.
van der Ven AT, Connaughton DM, Ityel H, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29:2348–2361.
van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29:36–50.
Lehmann OJ, Sowden JC, Carlsson P, Jordan T, Bhattacharya SS. Fox’s in development and disease. Trends Genet. 2003;19:339–344.
Kaufmann E, Knöchel W. Five years on the wings of fork head. Mech Dev. 1996;57:3–20.
Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241.
Fitch N, Kaback M. The Axenfeld syndrome and the Rieger syndrome. J Med Genet. 1978;15:30–34.
Nishimura DY, Swiderski RE, Alward WL, et al. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998;19:140–147.
Mears AJ, Jordan T, Mirzayans F, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld–Rieger anomaly. Am J Hum Genet. 1998;63:1316–1328.
Alward WL. Axenfeld–Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130:107–115.
Ma AS, Grigg JR, Jamieson RV. Phenotype–genotype correlations and emerging pathways in ocular anterior segment dysgenesis. Hum Genet. 2019;138:899–915.
Nishimura DY, Searby CC, Alward WL, et al. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001;68:364–372.
Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld–Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007;48:228–237.
Chakrabarti S, Kaur K, Rao KN, et al. The transcription factor gene FOXC1 exhibits a limited role in primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2009;50:75–83.
Churchill A, Booth A. Genetics of aniridia and anterior segment dysgenesis. Br J Ophthalmol. 1996;80:669–673.
Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006;51:213–231.
Gripp KW, Hopkins E, Jenny K, Thacker D, Salvin J. Cardiac anomalies in Axenfeld–Rieger syndrome due to a novel FOXC1 mutation. Am J Med Genet A. 2013;161:114–119.
Du R-F, Huang H, Fan L-L, Li X-P, Xia K, Xiang R. A novel mutation of FOXC1 (R127L) in an Axenfeld–Rieger syndrome family with glaucoma and multiple congenital heart diseases. Ophthalmic Genet. 2016;37:111–115.
Green MC. The developmental effects of congenital hydrocephalus (ch) in the mouse. Dev Biol. 1970;23:585–608.
Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996.
Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000;127:1387–1395.
Komaki F, Miyazaki Y, Niimura F, Matsusaka T, Ichikawa I, Motojima M. Foxc1 gene null mutation causes ectopic budding and kidney hypoplasia but not dysplasia. Cells Tissues Organs. 2013;198:22–27.
Motojima M, Tanimoto S, Ohtsuka M, Matsusaka T, Kume T, Abe K. Characterization of kidney and skeleton phenotypes of mice double heterozygous for Foxc1 and Foxc2. Cells Tissues Organs. 2016;201:380–389.
Braun DA, Sadowski CE, Kohl S, et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet. 2016;48:457–465.
Gee HY, Otto EA, Hurd TW, et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014;85:880–887.
Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–1289.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9.
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539
Genome Aggregation Database (gnomAD). Version 2.1. Cambridge, MA: The Broad Institute of MIT and Harvard. 2018. https://gnomad.broadinstitute.org/.
UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515.
Chapman CJ, Bailey RR, Janus ED, et al. Vesicoureteric reflux: segregation analysis. Am J Med Genet. 1985;20:577–584.
Noe HN, Wyatt RJ, Peeden JN, Rivas ML. The transmission of vesicoureteral reflux from parent to child. J Urol. 1992;148:1869–1871.
Warejko JK, Tan W, Daga A, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2018;13:53–62.
Grüneberg H. Congenital hydrocephalus in the mouse, a case of spurious pleiotropism. J Genet. 1943;45:1–21.
Nakano T, Niimura F, Hohenfellner K, Miyakita E, Ichikawa I. Screening for mutations in BMP4 and FOXC1 genes in congenital anomalies of the kidney and urinary tract in humans. Tokai J Exp Clin Med. 2003;28:121–126.
Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Meth Enzymol. 1996;266:554–571.
Acknowledgements
We thank the families and study individuals for their contribution. This research was supported by grants from the National Institutes of Health to F.H. (DK088767). F.H. is also supported by the Begg Family Foundation. C.-H.W.W. is supported by funding from the National Institutes of Health (grant T32-GM007748). N.M. is supported by funding from the National Institutes of Health (grant T32-DK007726). M.N. is supported by a grant from the Japan Society for the Promotion of Science. D.M.C. is funded by the Health Research Board, Ireland (HPF-206–674); the International Pediatric Research Foundation Early Investigators’ Exchange Program; and the Amgen Irish Nephrology Society Specialist Registrar Bursary. F.K. is supported by funding from the Biomedical Education Program. V.K. is supported by the Deutsche Forschungsgemeinschaft (403877094). We also thank the Yale Center for Mendelian Genomics for exome sequencing analysis (U54HG006504).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
F.H. is a cofounder and S.A.C. member and holds stock in Goldfinch-Bio. All other authors declare that they have no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, CH.W., Mann, N., Nakayama, M. et al. Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med 22, 1673–1681 (2020). https://doi.org/10.1038/s41436-020-0844-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0844-z
Keywords
This article is cited by
-
The genetics and pathogenesis of CAKUT
Nature Reviews Nephrology (2023)
-
A novel missense mutation of FOXC1 in an Axenfeld–Rieger syndrome patient with a congenital atrial septal defect and sublingual cyst: a case report and literature review
BMC Medical Genomics (2021)
