
Abstract
Pathogenic variants in the CFTR gene are causative of classic cystic fibrosis (CF) as well as some nonclassic CF phenotypes. In 2001, CF became the first target of pan-ethnic universal carrier screening by molecular methods. The American College of Medical Genetics and Genomics (ACMG) recommended a core panel of 23 disease-causing variants as the minimal set to be included in pan-ethnic carrier screening of individuals with no family history of the disease, and these variants were usually assessed using targeted methods. The original recommendation also left open the option for laboratories to offer expanded CFTR variant panels; however, at the time, expanded CFTR variant panels were met with some controversy on the basis of the available technologies and the limited phenotypic knowledge of rare variants. Both of those aspects have now evolved, prompting this update of the ACMG technical standards for CFTR variant testing.
Similar content being viewed by others

INTRODUCTION
It has now been 30 years since the discovery of the CFTR gene. Pathogenic variants in this gene are causative of classic cystic fibrosis (CF) as well as nonclassic CF phenotypes, including isolated congenital absence of the vas deferens (CAVD).1,2,3 Identification of the CFTR gene resulted in the availability of diagnostic testing for symptomatic individuals and carrier testing for at-risk relatives. CFTR variant testing is also now offered to reproductive couples in the United States and some other countries for screening purposes, is included in most newborn screening programs, and is being used for personalized therapy.
In 2001, upon joint recommendation by the American College of Medical Genetics and Genomics (ACMG) and the American College of Obstetricians and Gynecologists (ACOG), CF became the first target of pan-ethnic universal carrier screening by molecular methods. Because molecular genetic testing at the time was performed largely by targeted allele-specific assays, the ACMG recommended a core panel of 25 disease-causing variants with an allele frequency of ≥0.1% in the general US population to represent the minimal set to be included in pan-ethnic carrier screening of individuals with no family history of the disease.4 Aside from the elimination of two variants from the panel in 2004—one because it was much rarer than previously thought, and the other because it was determined to actually be a benign variant—this core panel (often referred to as the “ACMG-23”) has remained unchanged since, even as molecular diagnostic technologies and genetic knowledge have dramatically advanced.5
However, with the advent of next-generation sequencing (NGS), it has become much easier and less expensive to more extensively interrogate genes including all possible coding variants as well as deep-intronic and other noncoding variants if desired. Furthermore, our knowledge of additional CFTR variants at the functional level has invited a broadening of the scope of variation that could potentially be tested for, in either the carrier or diagnostic setting. The original ACMG-ACOG recommendation left open, but did not encourage, the option for laboratories to offer an expanded CFTR variant panel beyond the core panel, and at the time, expanded variant panels were met with some controversy on the basis of the available technologies and limited phenotypic knowledge of rare variants. However, both of those aspects have now evolved, prompting this update of the ACMG technical standards for CFTR variant testing.
BACKGROUND ON THE CFTR GENE
Gene symbol/chromosome locus
The CFTR (cystic fibrosis transmembrane conductance regulator) gene is located on chromosome 7q31.2 and contains 27 coding exons. The reference messenger RNA (mRNA) and protein sequences are NM_000492 and NP_000483, respectively.
MIM numbers
The MIM number for the CFTR gene is 602421, and variants within the CFTR gene are associated with autosomal recessive cystic fibrosis (MIM 219700). Variants within the CFTR gene are also associated with nonclassic CF phenotypes including autosomal recessive CAVD (MIM 277180), autosomal dominant idiopathic pancreatitis (MIM 167800), and autosomal dominant bronchiectasis (as a modifier) (MIM 211400).
Brief clinical description of the CFTR-associated phenotypes
Classic cystic fibrosis is one of the most common autosomal recessive conditions in Caucasians with a prevalence of approximately 1 in 2500 to 3300 live births. Classic CF is characterized by viscous mucus in the lungs along with involvement of the digestive system, reproductive system, and sweat glands. The degree of pulmonary disease determines the prognosis; recurrent and persistent pulmonary infections are common and lead to respiratory failure.
Pancreatic insufficiency occurs in cases of classic CF, and neonatal meconium ileus occurs in 15% to 20% of newborns with this form of CF. Other findings may include poor growth, poor weight gain despite normal food intake, salty skin, chronic sinusitis, nasal polyps, liver disease, pancreatitis, and CAVD. The overall average survival of classic CF patients, including those with a milder presentation, is approximately 40 years.
Nonclassic CF phenotypes include infertility, idiopathic pancreatitis, bronchiectasis, and chronic rhinosinusitis. The presence of pancreatic exocrine function and intermediate (<90 mmol/l) or normal (<40 mmol/l) chloride levels after employing sweat testing help to distinguish nonclassic from classic forms.6,7
CFTR gene description/normal gene product
The CFTR gene encodes an adenosine triphosphate (ATP)–binding cassette transporter protein. This protein functions as a channel that transports negatively charged chloride ions across the membranes of various cells that produce mucus, sweat, saliva, tears, and digestive enzymes. The transport of these ions helps control the flow of water in cells, so that the necessary mucus is made, which is used to lubricate the lining of several organs and tissues, including the lungs, digestive system, and reproductive system. The CFTR protein also influences the transport of positively charged sodium ions across cell membranes.
Disease mechanisms/abnormal gene product/treatment
Pathogenic variants in the CFTR gene alter the production, structure, or stability of the chloride channel. This impairs the transport of chloride ions and the movement of water into and out of cells, which results in the production of mucus that is abnormally thick and viscous by the cells lining the passageways of the lungs, pancreas, and other organs. Lung disease results from the clogging of the airways due to mucus build-up, decreased mucociliary clearance, and the resulting infection and inflammation. Thickened secretions from the pancreas block the exocrine movement of the digestive enzymes into the duodenum and result in irreversible damage to the pancreas, often with pancreatitis. The lack of digestive enzymes leads to malabsorption, which contributes to malnutrition and poor growth and development. Thickened secretions may also cause liver problems, which can lead to scarring and cirrhosis.
Symptomatic treatment for classic CF includes control of infections, clearance of mucus in the lung, and improvement of nutrition through pancreatic enzymatic replacement. The development of small molecule drugs that target the folding and functional defects associated with abnormal forms of CFTR have revolutionized the treatment of the disorder,8 and many individuals with classic CF will likely benefit from combinations of these newly developed CFTR-targeted drugs.9,10 Patients with nonclassic CF will present with variable phenotypes and disease severity (expressivity). Management of these patients will depend on disease manifestation and severity and are more likely to target the organ system involved (e.g., antibiotic therapy).
Indications for CFTR variant testing
CFTR variant testing is used postnatally for diagnosis and in adults for diagnosis and carrier testing. Diagnostic testing can be used for the molecular confirmation of a clinical CF diagnosis, for infants with meconium ileus, for males with CAVD, for individuals with idiopathic pancreatitis or bronchiectasis, and as a follow-up to newborn screening. Patients with classic CF are more likely to have two variants that together result in a complete loss of protein function, whereas those with nonclassic CF are more likely to have two variants that together result in only a partial loss of protein function. Carriers of a single pathogenic or likely pathogenic variant are expected to be asymptomatic, and couples can be tested sequentially (with the female typically tested first and the male tested only if the female carries a variant) or simultaneously. During pregnancy, simultaneous testing may be desired depending on gestational age, family and personal history, ethnicity, or patient preferences. Carrier testing may be offered to individuals with a positive family history of CF, in partners of individuals with a positive family history, in partners of CAVD males, to reproductive age women, and to gamete donors.
CFTR variant testing can also be performed for prenatal diagnosis using cells obtained for diagnostic cytogenetic testing (i.e., amniocentesis or chorionic villus sampling [CVS]). Testing can take place on cultured or uncultured amniocytes or villous trophoblasts.
TESTING CONSIDERATIONS (WHAT TO TEST)
Modernizing the ACMG-23 variant panel
The development of the ACMG-23 variant panel followed a careful analysis and revision of the original ACMG-25 variant panel, which was a product of two National Institutes of Health (NIH) consensus conferences (1997 and 1998), followed by a Steering Committee made up of ACMG and ACOG representatives.4,5 This was the first time professional organizations recommended population-based screening at the DNA level for a genetic condition.
However, along with advances in technology, the past two decades have brought about an improved understanding of genetics and genomics. As a result, (1) the system of variant classification has been refined, (2) the phenotypes associated with CF (both classic and nonclassic forms) have been better characterized, (3) the associations of CFTR variants with clinically relevant nonclassic CF phenotypes are now recognized, (4) in vitro genotype–phenotype functional variant analysis exists, and (5) pan-ethnic screening with minimal variation in implementation is accepted.
Expanded carrier screening by NGS now makes it possible to screen for clinically relevant variants without regard to ethnicity.11 The bottleneck is no longer the number of detectable variants but instead an improved understanding of genotype–phenotype correlation.
Classification-based (targeted) testing vs. classification-based reporting
As a way to ensure that CFTR variant testing for carrier screening and diagnostic testing purposes remains comprehensive, pan-ethnic, and up-to-date, the ACMG recommends either a classification-based reporting approach or a classification-based (targeted) testing approach (which has historically been used for CFTR carrier screening). For those laboratories who wish to continue using a targeted testing approach, the ACMG-23 variant panel remains as the minimum list of CFTR variants that should be included. Laboratories may want to consider adding additional variants to their panel depending on the ethnic composition of their expected test population.12 However, the minimum list of CFTR variants recommended for pan-ethnic carrier screening has not been increased at this time.
In contrast, the classification-based reporting approach is based on providing individuals with comprehensive CF testing that minimally includes an assessment of all of the exonic coding regions and +/−2 bp proximal splice junctions of the CFTR gene, followed by the reporting of all pathogenic and likely pathogenic variants for classic CF. Variants of uncertain clinical significance (VUS) are discussed in more detail in “Considerations for variants of uncertain clinical significance.” Variants that are benign or likely benign should not be reported. Targeted and comprehensive approaches are both acceptable for the testing of individuals regardless of race, ethnicity, or test indication.
The classification-based reporting approach is similar to the approach recommended for the reporting of secondary findings in the setting of genomic sequencing (i.e., exome and genome sequencing), whereby specific variant classifications/types in specific genes are recommended for reporting but specific variants are not recommended.13,14 Many laboratories also use a similar approach for the reporting of primary findings from diagnostic genomic sequencing, with pathogenic and likely pathogenic variants being reported. Further considerations on specific aspects of these approaches for CFTR testing are provided below.
Considerations for intronic variants
Aside from the important polyT/TG region in intron 9 (discussed in more detail in “Considerations for the intron 9 polyT and TG regions”) and the c.2657+5G>A (also known as 2789+5G>A) and c.3718–2477C>T (also known as 3849+10kbC>T) variants included in the original ACMG-23 variant panel, little attention thus far has been paid to deep-intronic variants in the CFTR gene, at least at the clinical level. Targeted variant testing would not typically interrogate such variants, nor would exome sequencing (other than the detection of potential splice variants surrounding the proximal intron–exon junctions). However, with genome sequencing likely to become more common in clinical molecular diagnostics in the coming years, it is inevitable that deep-intronic variants in CFTR will be increasingly observed and will need to be annotated. In most cases, the question to be answered will be whether the intronic variant creates a cryptic splice site leading to inclusion of a pseudoexon or loss of a true exon in the mRNA product of the gene, resulting in an abnormal CFTR protein.15 Despite the existence of splice variant prediction algorithms, the best confirmatory evidence would be obtained with RNA sequencing, but that approach may require the biopsy of a tissue in which CFTR is sufficiently expressed, and such invasive studies would not be practical in most clinical laboratory settings.16 In the absence of RNA studies, it is recommended that laboratories exercise caution in classifying and reporting deep-intronic variants, if detected.
As they were originally part of the ACMG-23 carrier screening variant panel, for all prenatal, postnatal, and adult diagnostic testing and carrier screening indications for CFTR sequencing, the ACMG recommends the reporting of the c.2657+5G>A and c.3718–2477C>T intronic variants. If these variants are not detectable with the laboratory’s methodology (e.g., exome sequencing), then a separate assay should be performed to test for these variants.
Considerations for deletions/duplications
Gene deletions/duplications may include a single exon, multiple exons, or the entire gene. In addition, some deletions may extend beyond the entire gene and have additional implications. While deletions and duplications occur in CFTR, their frequency is estimated at <5% of all detected CFTR variants, which may be an underestimate due to a historical lack of copy-number variant (CNV) data available from standard analyses.
For all prenatal, postnatal, and adult diagnostic testing and carrier screening indications for CFTR testing, the ACMG does not recommend the testing of any specific exon-level or gene-level deletion or duplication variants. However, as larger deletions and duplications are highly likely to be pathogenic if detected, the ACMG recommends that laboratories using classification-based reporting methods should be capable of evaluating for the presence of deletions and duplications in prenatal and postnatal diagnostic indications, especially when only a single variant is initially detected. If a laboratory does not currently have the capability to detect deletions and duplications using their primary testing methodology, it is recommended that laboratories validate their ability to detect deletions and duplications using an alternate methodology or assist in arranging for suitable send-out testing to be ordered by clinicians when needed.
METHODOLOGY CONSIDERATIONS (HOW TO TEST)
All general recommendations described in the ACMG Technical Standards for Clinical Genetics Laboratories apply. The following additional details are specific for CFTR variant testing.
Positive controls
Positive controls for many CFTR variants can be obtained from the National Institute of General Medical Sciences (NIGMS) Human Genetic Cell Repository as either cell lines or DNA. Synthetic super controls that include multiple variants are commercially available.
Sample preparation
CFTR variant testing typically occurs using genomic DNA prepared from whole blood using a variety of automated and manual extraction methods. Other extraction methods can also be used that accommodate buccal samples (e.g., brushes, swabs), saliva, and prenatal specimens (e.g., amniocytes, chorionic villi).
Method verification/validation
For CFTR variant analysis, laboratories can choose between the creation of a laboratory developed test (LDT) or the use of an existing in vitro diagnostic (IVD) (i.e., an FDA-cleared or approved test). Laboratories offering genetic tests for clinical purposes are regulated under CLIA. IVD tests require a method verification by the performing laboratory, whereas LDTs require a more comprehensive method validation.
Targeted vs. comprehensive methods
Historically, a variety of molecular techniques were used to identify various types and numbers of CFTR variants, and the identification of CFTR variants could be broadly divided into two categories: (1) methods that targeted the detection of known variants, and (2) more comprehensive methods that attempted to detect all variants without a need for any prior knowledge regarding the identity or precise location of any particular variant. The specific testing strategy utilized by a laboratory often involved one or a combination of both types of methods (e.g., reflex testing), and laboratories were aware of the limitations of the method(s) chosen.
The ACMG recommends that laboratories performing initial CFTR variant testing on an individual can use either targeted or comprehensive methods to evaluate the gene. The testing methodology chosen by a laboratory for a specific indication, including the corresponding limitations, should be clearly communicated in the report. If pathogenic or likely pathogenic CFTR variants have been confirmed in both biological parents, or an affected full sibling, only targeted methods should be used. Several targeted methods are described in detail in the ACMG Technical Standards for Clinical Genetics Laboratories. Information regarding some comprehensive methods for CF testing are described further below.
Comprehensive methods for the detection of CFTR variants
Sanger sequencing
Sanger sequencing has been routinely used in clinical laboratories for the analysis of CFTR due to its robustness, accuracy, and ease of set-up. Sanger sequencing protocols for the analysis of CFTR, including the 5′-flanking region, 27 coding exons, and selected intronic regions, have previously been published.17 Sanger sequencing can also be used for targeted testing of specific familial variants that have previously been identified. However, some important limitations related to the Sanger sequencing of CFTR include low throughput, the possibility of allele dropout and false-negative results, and an inability to detect large deletions or duplications.
Next-generation sequencing
NGS technologies have revolutionized the genetic testing field with a substantial reduction in cost per base and a considerable enhancement of sequencing capacities, allowing for the simultaneous analysis of single genes or panels of selected genes in a large number of patients at a previously unprecedented speed and cost.18 NGS-based protocols for the analysis of CFTR have demonstrated acceptable results; however, several drawbacks have remained, such as a risk of false-negative or false-positive results and the challenge of sequencing specific regions (often requiring additional Sanger sequencing to adequately assess those regions).
Deletions and duplications can also be analyzed from NGS data using normalized depth of coverage and paired-end mapping, and a workflow for CFTR variant analysis using NGS for the simultaneous detection of single-nucleotide variants (SNVs) and large rearrangements has also previously been published.19 While NGS is currently more time- and cost-effective than running a separate companion assay for dosage analysis, the sensitivity and specificity of the bioinformatics analysis for large deletions and duplications depends highly on the quality of NGS data produced.
Multiplex ligation-dependent probe amplification (for deletion/duplication testing)
Multiplex ligation-dependent probe amplification (MLPA) is a polymerase chain reaction (PCR)–based method for quantifying multiple genomic loci in a single reaction. It is based on the ligation of a set of two oligonucleotide probes that have annealed adjacently to a target sequence, and only ligated probes can serve as a template for a subsequent PCR. MLPA is an efficient method to detect large deletions and duplications in the CFTR gene, and commercial reagents exist.20 A limitation of MLPA is that any DNA sequence variants located directly under probe binding sites may interfere with probe hybridization and could result in false-positive carrier (one copy) or diagnostic (zero copies) results.
Exon-targeted array CGH (for deletion/duplication testing)
High-resolution array comparative genomic hybridization (aCGH) with oligonucleotide probes densely distributed across individual genes can be developed to detect copy-number changes at the single-exon level. Oligonucleotide probes can be designed in silico for any sequenced region of a genome, thus allowing the flexibility of interrogating any region of interest. An exon-targeted aCGH for the detection of intragenic CFTR CNVs has been described.21 One advantage of aCGH over MLPA (or other PCR-based methods) is that whenever a deletion or duplication event is identified, its breakpoints can be narrowed down to more precise genomic locations. This greatly facilitates the effort of breakpoint characterization at the DNA sequence level. Furthermore, the high probe density across the gene serves to avoid the pitfalls of false-positive single-exon losses that can result from DNA sequence variants affecting primer or probe binding sites.
REPORTING CONSIDERATIONS (WHAT TO REPORT)
Considerations for variant nomenclature/classification
The use of standard Human Genome Variation Society (HGVS) nomenclature is required for the accurate communication of genetic testing results to health-care providers.22 However, for conditions such as CF, laboratory geneticists and clinicians may still wish to use historical, common terminology (such as deltaF508) in their reports and clinical notes, and it is currently acceptable to describe a CFTR variant using historical nomenclature as long as standard HGVS nomenclature is used as well.
Regardless of the test indication, all CFTR variants should be classified using ACMG sequence variant classification recommendations.23 Information from CFTR variant–containing databases can be used to inform those variant classifications.
Considerations regarding the use of CFTR variant–containing databases
CFTR2
The Clinical and Functional Translation of CFTR (CFTR2) project was initiated in 2008 to expand the clinical annotation of CFTR variants beyond the original ACMG-23 variant panel. However, the utility of genetic testing for the diagnosis of, and therapy for, CF has expanded the goal of the CFTR2 project to encompass all variants reported in CFTR.24 Of ~2000 variants reported in the CF Mutation Database (www.genet.sickkids.on.ca), >1600 occur in the >89,000 subjects participating in the CFTR2 project.
Annotation of CFTR variants by the CFTR2 team is performed using a three-level approach: clinical criteria, functional assessment, and penetrance analysis. Once variants have been assessed, results are uploaded to the CFTR2 website (CFTR2.org) so that interpretations are immediately available to the public. Variant assessments within the CFTR2 project align well with assignments of disease liability using the ACMG variant classification recommendations.25 Since the selection of variants for annotation is based on worldwide frequency, rather than country, ethnic, or racial frequencies, variant information in CFTR2 is applicable to all populations. The pan-ethnic nature of these data enable interpretation of assays that interrogate the entire coding region of CFTR in individuals regardless of race or ethnicity.
While it is possible to indicate that certain variants do not cause CF, it does not exclude the possibility that they alter CFTR function and contribute to a “CFTR-related disorder” (i.e., male infertility, pancreatitis). Furthermore, the CFTR2 project is unable to assign a small subset of variants (approximately 5%) to disease or non-disease-causing categories due to incongruities among clinical, function, or penetrance data. Some of these variants are classified as variants of varying clinical consequence by the CFTR2 project, and they have been reported both in individuals with CF as well as in healthy individuals (in trans with a known disease-causing CFTR variant in each situation). The variants of varying clinical consequence are often associated with functional thresholds that allow genetic and environmental modifiers to determine whether an individual manifests CF.25 These observations also help to substantiate reporting VUS (see “Considerations for variants of uncertain clinical significance”).
ClinVar
ClinVar (www.ncbi.nlm.nih.gov/clinvar) contains CFTR variants that have been expertly curated by the CFTR2 project, variants that are generally rare and have been evaluated by clinical molecular diagnostic laboratories, and variants that have been reported by researchers. The level of confidence for the disease annotation assigned to variants outside of the CFTR2 project varies. A small fraction of rare variants have been reported by multiple laboratories and have the same assertion of pathogenicity, lending a degree of confidence to these assignments. However, for other variants, only a single laboratory has provided an assertion or multiple laboratories provided different variant classifications. In most cases, variants reported outside of the CFTR2 project have not had in-depth functional assessment, and in an unknown fraction of cases, the diagnosis of CF has not been confidently confirmed. Therefore, laboratories should exercise caution when evaluating non-CFTR2 assertions for CFTR variants within ClinVar.
Considerations for the determination of variant phasing (cis vs. trans, de novo)
For many rare recessive disorders, the finding of two potentially pathogenic variants in an affected individual requires the determination of whether they are in cis (on the same chromosome) or in trans (on different homologous chromosomes) to clarify the diagnosis. Parental testing is typically recommended in this setting to assess the origin of each of the variants so that targeted testing can be offered to other family members who wish to know their carrier status.
For CF, finding two well-described pathogenic variants (those with an allele frequency of ≥0.01% in the CFTR2 database) in an affected individual may not require additional testing to support the diagnosis. However, finding a rare novel CFTR variant in combination with a well-established pathogenic CFTR variant in an affected individual would ideally result in the ascertainment of both parents to determine the phase and confirm the diagnosis. A rare variant in an affected individual may also be de novo, though de novo events are rarely observed in individuals with CF.
If it is not possible to obtain samples from both parents and/or additional family members to determine the phase, caution should be used in the interpretation of the results. Laboratories should have a policy for follow-up familial testing, including what types of samples they will accept, and who can provide those samples.
Considerations for variants of uncertain clinical significance
As more individuals undergo comprehensive sequencing, the possibility of finding genetic variants with uncertain clinical significance increases. VUS arise from an inability to formally associate a variant and phenotype, which can be due to reduced penetrance, variable expressivity due to modifier genes and/or environmental factors, pleiotropy associated with variants in different regions of the same gene, as well as the possibility that the variant is non-disease-associated and is benign.
More than 2000 variants within CFTR have already been identified; however, most of the private or very rare variants are currently classified as VUS. Laboratories routinely reassess VUS to seek a more definitive classification (benign, likely benign, likely pathogenic, pathogenic), and unlike pathogenic and benign variants, the VUS classification is generally not static.26 Furthermore, genotype–phenotype correlations exist on a spectrum, and age-related penetrance of nonclassic phenotypes is common.
Some concerns surrounding the appropriateness of reporting VUS to health-care providers include unnecessary anxiety for the patient and the possibility for them to make reproductive decisions (e.g., pregnancy termination) based on misinformation. On the other hand, autonomy reflects the right of patients to know, become educated, and make their own decisions.
For all prenatal, postnatal, and adult diagnostic testing indications for CFTR where comprehensive methods are used, the ACMG recommends the reporting of VUS. For all adult carrier screening indications for CFTR where comprehensive methods are used, VUS should generally not be reported. However, laboratories may want to consider reporting VUS in the partner of an individual who had a pathogenic or likely pathogenic variant detected during screening.
Considerations for variants associated with variable expressivity
Some CFTR variants are associated with variable expressivity (these are referred to in the CFTR2 database as variants of varying clinical consequence). The phenotypes that may be associated with one of these variants when in trans with another one of these variants (or even when in trans with a known pathogenic variant) cannot often be predicted, and therefore results involving these variants should be interpreted carefully and reported thoughtfully.
For all prenatal, postnatal, and adult diagnostic testing indications for CFTR where comprehensive methods are used, the ACMG recommends the reporting of any variants associated with variable expressivity. In addition to the corresponding ACMG variant classification, if a variant has been classified by the CFTR2 project as a variant of varying clinical consequence, this terminology should also be included in the report. For all adult carrier screening indications for CFTR where comprehensive methods are used, variants associated with variable expressivity that are not currently classified as a pathogenic or likely pathogenic variant by the laboratory should generally not be reported. Laboratories may want to consider reporting variants associated with variable expressivity in the partner of an individual who had a pathogenic or likely pathogenic variant detected during screening.
The specific R117H variant and associated polyT tract is described further below.
Considerations for the intron 9 polyT and TG regions
The c.350G>A (p.Arg117His, commonly referred to as R117H) CFTR variant is associated with variable expressivity, and its consequence is dependent on the status of the polythymidine (polyT) tract in intron 9 (c.1210–12T[5], c.1210–12T[7], or c.1210–12T[9]).
For R117H/5T individuals, additional testing is needed to determine the phase of these variants, though sometimes phase can be inferred for a variant with a well-known polyT haplotype present on the opposite allele. For example, c.1521_1523delCTT (p.Phe508del) and 9T occur almost completely in cis. R117H and 5T in cis with a known CF-causing variant on the opposite allele generally results in nonclassic CF. R117H in cis with 7T variant and a CF-causing variant in trans may have a phenotype varying from asymptomatic to CAVD (in males) and very rarely to nonclassic CF.
Independent of R117H, the 5T allele by itself is associated with variable penetrance for CF and CAVD based on the status of an adjacent poly TG tract, which usually contains 11, 12, or 13 repeats (c.1210–34TG[11], c.1210–34TG[12], c.1210–34TG[13]). When paired with a known CF-causing variant, 5T and 11TG variants in cis rarely confer an increased risk for CAVD in males while 5T in cis with 12TG or 13TG confers risk for CAVD and rarely for nonclassic CF. Given the commonness of the 5T allele (one in ten individuals carry a 5T variant), interpretation of its disease liability should ideally be performed in the context of the number of associated TG repeats.
For all prenatal, postnatal, and adult diagnostic testing indications for CFTR, the ACMG recommends the reporting of R117H status as well as the results from at least the associated polyT tract. For all adult carrier screening indications for CFTR, polyT status should be reported when the R117H variant is detected; laboratories may also want to consider reporting the results from the associated polyT tract in the partner of an individual who had a pathogenic or likely pathogenic variant detected during screening.
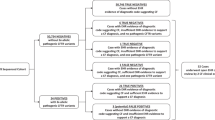
Additional considerations for prenatal diagnostic testing
When indicated in the prenatal setting:
-
1.
Targeted sequencing for specific CFTR variants may be considered when
-
(a)
A pathogenic or likely pathogenic variant is confirmed in both partners.
-
(b)
A pathogenic or likely pathogenic variant is confirmed in one partner and a VUS or variant associated with variable expressivity is confirmed in the other partner.
-
(c)
As part of preimplantation genetic testing when both biological parents are confirmed carriers of a pathogenic or likely pathogenic variant.
-
(a)
-
2.
Comprehensive CFTR sequencing may be considered when
-
(a)
One member of a couple is known to be a carrier of a pathogenic or likely pathogenic variant and any of the following is also true:
-
i.
The partner is unavailable for screening;
-
ii.
The partner has not been screened and any of the following is also true:
-
1.
Screening that partner would be cost prohibitive;
-
2.
The results from the partner would not be available in time to allow for reproductive decision making;
-
3.
A diagnostic procedure (e.g., CVS, amniocentesis) is also being performed for other reasons (e.g., ultrasound abnormality).
-
1.
-
i.
-
(b)
An ultrasound finding (i.e., fetal echogenic bowel) suggests an affected fetus and CFTR variant information is not available from either biological parent.
-
(a)
Quality assurance
Orthogonal confirmation
Please refer to other ACMG documents regarding general recommendations for the confirmation of germline variants that were originally detected using next-generation sequencing; Sanger sequencing is a commonly used method for this purpose.18 Orthogonal confirmation is not typically employed when methods other than NGS are used for initial CFTR testing. Laboratories should determine during the course of test validation whether orthogonal confirmation is required for the reporting of certain variants and what criteria will be used to make that determination. It is recommended that CFTR reports based on NGS methods clearly state whether or not a reported variant was confirmed using an alternate methodology; if orthogonal confirmation is not employed, a brief description of the criteria used to make that determination in the methods section of the report is recommended.
Variant reevaluation
Existing variant classifications may require modification as new evidence emerges. Please refer to the corresponding ACMG documents on the responsibilities of the laboratory and clinician regarding variant reevaluation.26,27
CONCLUSION
The ACMG recognizes that the detection capabilities of laboratories for SNVs and small insertions and deletions within the coding regions of genes are no longer a limiting factor for testing. The classification-based reporting approach is already used for diagnostic testing of many conditions, with pathogenic variants, likely pathogenic variants, and some VUS being reported. While these technical standards specifically address CFTR and its associated phenotypes, a classification-based reporting approach could eventually also be applied to other genes for carrier screening. However, there may be more VUS present in other genes, as many of the variants detected in other genes may not have been as extensively evaluated as the variants detected in CFTR.
In the future, laboratories may want to further consider the potential benefits (and potential negative impacts) of reporting all CFTR VUS for carrier screening. The joint interpretive capabilities of clinical laboratories and medical providers are expected to improve over time as more variants are routinely evaluated within the context of specific patient presentations and familial situations, ultimately leading to a greater degree of shared decision making between the patient and provider.
References
Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080.
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073.
Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065.
Grody WW, Cutting GR, Klinger KW, et al. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 2001;3:149–154.
Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–391.
Groman JD, Karczeski B, Sheridan M, et al. Phenotypic and genetic characterization of patients with features of "nonclassic" forms of cystic fibrosis. J Pediatr. 2005;146:675–680.
Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002;347:401–407.
Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56.
Davies JC, Moskowitz SM, Brown C, et al. VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379:1599–1611.
Keating D, Marigowda G, Burr L, et al. VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379:1612–1620.
Beauchamp KA, Johansen Taber KA, Grauman PV, et al. Sequencing as a first-line methodology for cystic fibrosis carrier screening. Genet Med. 2019;21:2569–2576.
Schrijver I, Pique L, Graham S, Pearl M, Cherry A, Kharrazi M. The spectrum of CFTR variants in nonwhite cystic fibrosis patients: implications for molecular diagnostic testing. J Mol Diagn. 2016;18:39–50.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574.
Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SFv2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255.
Lee M, Roos P, Sharma N, et al. Systematic computational identification of variants that activate exonic and intronic cryptic splice sites. Am J Hum Genet. 2017;100:751–765.
Bergougnoux A, Délétang K, Pommier A, et al. Functional characterization and phenotypic spectrum of three recurrent disease-causing deep intronic variants of the CFTR gene. J Cyst Fibros. 2019;18:468–475.
Strom CM, Huang D, Chen C, et al. Extensive sequencing of the cystic fibrosis transmembrane regulator gene: assay validation and unexpected benefits of developing a comprehensive test. Genet Med. 2003;5:9–14.
Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747.
Pagin A, Devos A, Figeac M, et al. Applicability and efficiency of NGS in routine diagnosis: in-depth performance analysis of a complete workflow for CFTR mutation analysis. PLoS One. 2016;11:e0149426.
Schrijver I, Rappahahn K, Pique L, Kharrazi M, Wong LJ. Multiplex ligation-dependent probe amplification identification of whole exon and single nucleotide deletions in the CFTR gene of Hispanic individuals with cystic fibrosis. J Mol Diagn. 2008;10:368–375.
Saillour Y, Cossée M, Leturcq F, et al. Detection of exonic copy-number changes using a highly efficient oligonucleotide-based comparative genomic hybridization-array method. Hum Mutat. 2008;29:1083–1090.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45:1160–1167.
Raraigh KS, Han ST, Davis E, et al. Functional assays are essential for interpretation of missense variants associated with variable expressivity. Am J Hum Genet. 2018;102:1062–1077.
Deignan JL, Chung WK, Kearney HM, et al. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:1267–1270.
David KL, Best RG, Brenman LM, et al. Patient re-contact after revision of genomic test results: points to consider–a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:769–771.
Author information
Authors and Affiliations
Consortia
Ethics declarations
Disclosure
J.L.D., C.A., D.d.G., W.W.G., K.G.M., and S.R. all serve as directors in clinical laboratories that perform a breadth of genetic and genomic analyses on a fee-for-service basis. K.G.M. is an employee of GeneDx, a wholly owned subsidiary of OPKO Health, Inc. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer:
This technical standard is designed primarily as an educational resource for clinical laboratory geneticists to help them provide quality clinical laboratory genetic services. Adherence to this technical standard is voluntary and does not necessarily assure a successful medical outcome. This technical standard should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinical laboratory geneticist should apply his or her own professional judgment to the specific circumstances presented by the individual patient or specimen.
Clinical laboratory geneticists are encouraged to document in the patient’s record the rationale for the use of a particular procedure or test, whether or not it is in conformance with this technical standard. They also are advised to take notice of the date any particular technical standard was adopted, and to consider other relevant medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this technical standard on 16 March 2020.
Rights and permissions
About this article

Cite this article
Deignan, J.L., Astbury, C., Cutting, G.R. et al. CFTR variant testing: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 22, 1288–1295 (2020). https://doi.org/10.1038/s41436-020-0822-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0822-5
Keywords
This article is cited by
-
Diversity of CFTR variants across ancestries characterized using 454,727 UK biobank whole exome sequences
Genome Medicine (2024)
-
Clinical and genetic features of cystic fibrosis in Japan
Journal of Human Genetics (2023)
-
Ethical considerations in gene selection for reproductive carrier screening
Human Genetics (2022)
-
CFTR modulates aquaporin-mediated glycerol permeability in mouse Sertoli cells
Cellular and Molecular Life Sciences (2022)
-
Methods and feasibility study for exome sequencing as a universal second-tier test in newborn screening
Genetics in Medicine (2021)
