
Abstract
Purpose
Identifying rare genetic causes of common diseases can improve diagnostic and treatment strategies, but incurs high costs. We tested whether individuals with common disease and low polygenic risk score (PRS) for that disease generated from less expensive genome-wide genotyping data are more likely to carry rare pathogenic variants.
Methods
We identified patients with one of five common complex diseases among 44,550 individuals who underwent exome sequencing in the UK Biobank. We derived PRS for these five diseases, and identified pathogenic rare variant heterozygotes. We tested whether individuals with disease and low PRS were more likely to carry rare pathogenic variants.
Results
While rare pathogenic variants conferred, at most, 5.18-fold (95% confidence interval [CI]: 2.32–10.13) increased odds of disease, a standard deviation increase in PRS, at most, increased the odds of disease by 5.25-fold (95% CI: 5.06–5.45). Among diseased patients, a standard deviation decrease in the PRS was associated with, at most, 2.82-fold (95% CI: 1.14–7.46) increased odds of identifying rare variant heterozygotes.
Conclusion
Rare pathogenic variants were more prevalent among affected patients with a low PRS. Therefore, prioritizing individuals for sequencing who have disease but low PRS may increase the yield of sequencing studies to identify rare variant heterozygotes.
Similar content being viewed by others

INTRODUCTION
Most common diseases are at least partially heritable1 and can sometimes be due to rare pathogenic variants.2 Identifying rare variant heterozygotes can sometimes change clinical care by tailoring diagnostic and/or treatment strategies.3 For example, treatment can be improved in individuals carrying rare pathogenic variants since they may respond more appropriately to specific therapies, such as in the case of monogenic forms of type 2 diabetes, where oral pills can be used instead of insulin therapy.4
Despite these advantages, rare variants are not routinely sought in the course of clinical care for most common diseases, often because targeted sequencing panels or exome sequencing are required. These are costly technologies that generally have low yield due to the rarity of actionable genetic variants in the population. Thus, efficient ways to triage individuals with common diseases for rare actionable variants could help to improve clinical care by identifying a group of individuals more likely to harbor rare causal genetic variants, who could then undergo required sequencing.
One way to effectively triage individuals for sequencing studies could be through the use of polygenic risk scores (PRS). This is because the heritable predisposition to any disease could arise through many common genetic variants of small effect (which are captured by PRS), or rare variants of large effect, or a combination of the two. That is, individuals with a disease but with low polygenic predisposition may be more likely to harbor a rare genetic variant of large effect. Further, rare genetic variants are not generally captured in PRS and are generally not in linkage disequilibrium (LD) with common variants.2 Thus, PRS are likely independent of rare variant heterozygote status.
Recently, large cohort resources have improved the genomic prediction of common diseases through PRS,5,6,7 which capture information from many single-nucleotide polymorphisms (SNPs) assayed from genome-wide genotyping. PRS assess common genetic variations in millions of SNPs and cost approximately $40 in a research context.8 Given these advantages, many large health-care systems have initiated research programs by genome-wide genotyping of a large proportion of their population.9,10,11 If genome-wide genotyping becomes more routine in the course of clinical care, this single investment could be used to generate PRS for several common diseases. Individuals with these common diseases but with low polygenic risk could then be considered for sequencing studies.
Here we test the hypothesis that individuals with a low polygenic risk for common disease are more likely to harbor rare genetic variants than individuals without a low polygenic risk. To do so, we generated PRS for five complex diseases: breast cancer, colorectal cancer, type 2 diabetes, osteoporosis, and short stature. We then assessed the frequency of rare causal genetic variants among 44,550 individuals with exome sequencing data from UK Biobank by different degrees of polygenic risk.
MATERIALS AND METHODS
Ethics statement
Ethics approval for the UK Biobank study was obtained from the North West Centre for Research Ethics Committee (11/NW/0382).9 The UK Biobank ethics statement is available at https://www.ukbiobank.ac.uk/the-ethics-and-governance-council/. All UK Biobank participants provided informed consent at recruitment. This research has been approved by the UK Biobank Board & Access Sub-Committee and conducted using the UK Biobank Resource under application number 27449. No patients or members of the general public were directly involved in the design, recruitment, or conduct of the study. After publication, dissemination of the results will be sought across different countries involving respective patient organizations, the general public, and other stakeholders; typically, across social media, scientific meetings, and media interviews.
Study cohort
We used the UK Biobank, one of the largest health cohort studies date, to maximize statistical power. During 2006–2010, the UK Biobank recruited more than 500,000 participants aged between 40 and 69 years based on multiple assessment centers located in the United Kingdom, and collected a wide range of phenotypes and biological samples.9 Though reported to be generally healthier, less obese, and less likely to smoke and drink alcohol,12 these participants are still widely considered representative of the general, especially white British population, in the United Kingdom. The UK Biobank conducted high-quality genome-wide genotyping and genotype imputation to the reference panel from the Haplotype Reference Consortium.9 Among 488,363 genotyped participants, 49,908 underwent exome sequencing.9 In this study, we focused on 440,346 participants of white British ancestry, including 44,550 exome-sequenced participants. We split this white British cohort into three data sets: (1) a training set including 385,905 participants (97.5% of the non-exome-sequenced individuals) to construct PRS, (2) a model selection set including 9891 participants (2.5% of the non-exome-sequenced) for tuning model parameters in the PRS, and (3) a test set including all 44,550 exome-sequenced participants which permitted identification of rare pathogenic variants. Demographic characteristics of these three subcohorts are provided in Table S1. In addition, the UK Biobank also genotyped approximately 50,000 participants with nonwhite British ancestries, including 5,358 participants who also underwent exome sequencing. The generalizability of our proposed principle in different ethnic groups was assessed based on these participants (see “Sensitivity analyses”).
Five diseases were examined: breast cancer, colorectal cancer, type 2 diabetes, osteoporosis, and short stature. Case status was defined using a combination of self-reported physician-made diagnoses and the International Classification of Diseases (ICD)-10 codes. Specifically, the ICD codes used were C50 (malignant neoplasm of breast) for breast cancer; C18 (malignant neoplasm of colon), C19 (malignant neoplasm of rectal sigmoid junction), or C20 (malignant neoplasm of rectum) for colorectal cancer; E11 (non-insulin-dependent diabetes mellitus), E13 (other specified diabetes mellitus), or E14 (unspecified diabetes mellitus) for non–type 1 diabetes; and M80 (osteoporosis with pathological fracture) or M81 (osteoporosis without pathological fracture) for osteoporosis. ICD-10 codes for cancer diagnoses were retrieved by the UK Biobank through the national cancer registries. Short stature was defined as having a normalized standing height lower than 75% of the population, after adjusting for age, sex, recruitment center, genotyping array, and the first 20 genetic principal components.13 This threshold for short stature was relaxed compared with most clinical definitions (2 standard deviations below the mean, representing ~2.3% of the general population14) to improve statistical power in our study.
Identification of heterozygotes of rare pathogenic variants
Among the 44,550 exome-sequenced participants, we identified clinically actionable genes following the guidelines of American College of Medical Genetics and Genomics (ACMG)15 or disease-causing genes with a dominant inheritance pattern validated in existing studies.16,17,18,19 These genes are BRCA1 and BRCA2 (both clinically actionable for hereditary breast cancer, as per the ACMG15) for breast cancer; MLH1, MSH2, MSH6, and PMS2 (all clinically actionable for hereditary nonpolyposis colorectal cancer, as per the ACMG15) for colorectal cancer; GCK, HNF1A, HNF1B, and HNF4A for monogenic diabetes;16 COL1A1, COL1A2, IFITM5, and PLS3 for osteoporosis;17 and SHOX, ACAN, NPR2, NPPC, and IHH for short stature.18,19
Variants affecting the clinically actionable or disease-causing genes above were considered candidate pathogenic variants. To remove potential false positive rare variants, we further required rare pathogenic variants to have a minor allele frequency (MAF) <0.1% in this population, a “high” or “moderate” functional impact predicted by SnpEff version 4.3,20 as well as a scaled Combined Annotation-Dependent Depletion (CADD) score >30 representing a higher pathogenicity than 99.9% of all variants, annotated by CADD version 1.5.21 A complete list of rare pathogenic variants identified for each disease is provided in Table S2. Individuals carrying at least one rare pathogenic variant of the associated disease were considered heterozygotes.
Construction of PRS
We constructed PRS leveraging SNPs with a MAF ≥0.1% and an imputation quality (INFO) score >0.3 following optimized strategies in existing studies for each disease.
For breast cancer, we directly employed a powerful and widely validated PRS consisting of 313 common variants derived from 69 European prospective studies.22
For type 2 diabetes, osteoporosis, and short stature diseases, we began by performing de novo genome-wide association studies (GWAS). For osteoporosis, we used a measure of bone strength, speed of sound (SOS) from ultrasound, which is a measure of directly related to the risk of osteoporosis23 and for which we have previously generated a PRS called gSOS (genetically predicted SOS).24 For short stature, we used standing height. The GWAS were run using linear mixed models adjusted for age, sex, recruitment center, genotyping array, and the first 20 genetic principal components in our training data set. We then undertook meta-analysis of our GWAS summary statistics for height with those obtained by the Genetic Investigation of ANthropometric Traits (GIANT) consortium25 to increase generalizability. A PRS for type 2 diabetes was constructed on the training set using LDPred26 wherein the proportion of causal markers (ρ) was optimized on the model selection set to be 0.01, following previous work from Khera et al.5 Following Forgetta et al.,24 the PRS for SOS and normalized height were constructed on the training set using L1-penalized least absolute shrinkage and selection operator (LASSO) regression, and the penalty parameters (λ) were optimized on the model selection set to be 5×10−4 for the SOS PRS and 5×10−2 for the height PRS, respectively.
Finally, all PRS were standardized to have zero mean and unit standard deviation in the test set. We reversed the sign of the PRS for SOS because a low predicted SOS would be indicative of a high risk of osteoporosis.23,24
For colorectal cancer, we retrieved 54 independent SNPs identified by Weigl et al. with known risk association among European descendants.27 Since the original study relied on only 1043 participants,27 the estimated effect sizes of these SNPs might have limited accuracy. Therefore, we performed multivariate logistic regression adjusted for age and sex on the training set.28 Coefficients for these SNPs obtained in this model were then used to derive a PRS for each individual in the test set.
Association of rare variants and the PRS with disease status
We first tested the marginal association between carrying rare pathogenic variants and the prevalence of disease by Fisher’s exact test. Logistic regressions adjusted for age and sex were adopted to test whether a high polygenic predisposition (that is, a higher PRS score) conferred a higher risk of the corresponding diseases. We next tested whether the mean and the distribution of PRS were significantly different between heterozygotes of rare pathogenic variants and nonheterozygotes, by Student t-tests and Kolmogorov–Smirnov (KS) tests, respectively. A joint logistic regression model adjusted for age and sex was used to test whether the rare and common risk components modify the respective magnitude of association with disease outcome. Finally, among diagnosed patients of each disease, we tested by logistic regression whether rare pathogenic variants were more prevalent among individuals with a low polygenic predisposition. All statistical analyses were conducted using R version 3.6.1.
Sensitivity analyses
We tested whether including other potentially causal genes could reinforce or weaken the associations we observed. We included rare pathogenic variants defined using the criteria above that affected TP53 (clinically actionable for Li–Fraumeni syndrome15), PALB2 (established causal gene for hereditary breast cancer29), and ATM (widely adopted in breast cancer gene panel sequencing30 with strong link to breast cancer31) for breast cancer, as well as those affecting APC (clinically actionable for familial adenomatous polyposis15), STK11 (clinically actionable for Peutz–Jeghers syndrome15), BMPR1A, and SMAD4 (both clinically actionable for juvenile polyposis15) for colorectal cancer.
We also examined how dependent our results were on the pathogenicity of rare variants by relaxing the threshold on scaled CADD score to 20, representing a higher pathogenicity than 99% of all variants. We repeated testing for associations between carrying rare pathogenic variants and developing corresponding diseases, and between rare and common genetic causes among diagnosed patients.
Further, we evaluated whether our findings could be replicated among 5,358 UK Biobank participants of six different meta-nonwhite British ancestries that underwent exome sequencing (Table S3). Because the nonwhite British populations had limited sample sizes to reliably derive population-specific MAFs, identification of rare pathogenic variant heterozygotes was based on Table S2.
RESULTS
Rare pathogenic variants conferred increased risk for corresponding diseases
The 440,346 white British participants in the UK Biobank were randomly split into three subcohorts of similar demographic characteristics for PRS development, model selection, and evaluation (“Materials and methods”; Table S1). The least prevalent disease under investigation, colorectal cancer, affected at least 0.9% of population. In the exome-sequenced white British cohort (N = 44,550), we identified 50 (0.21%) women carrying rare pathogenic variants affecting BRCA1 or BRCA2, as well as 199 (0.45%), 47 (0.11%), 189 (0.43%), and 139 (0.32%) individuals carrying rare pathogenic variants in causal genes of colorectal cancer, monogenic diabetes, osteoporosis, and short stature, respectively.
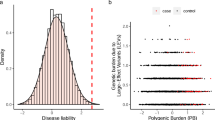
Heterozygotes of rare pathogenic variants were at 4.83-fold (95% confidence interval [CI]: 2.06–10.12; p value = 2.8×10−4) increased risk for breast cancer and 5.18-fold (95% CI: 2.32–10.13; p value = 1.1×10−4) increased risk for colorectal cancer (Fig. 1). Carrying rare pathogenic variants also seemed to confer increased risk, at a lower magnitude, for type 2 diabetes, osteoporosis, and short stature (Fig. 1a); for osteoporosis, the odds ratio (OR) was not significantly different from the null, but only four cases carried a rare pathogenic variant.

Carrying rare pathogenic variants was associated with increased risk of developing diseases. Nonheterozygotes were considered as the reference group for assessing the effect of rare pathogenic variants. Odds ratio associated with per–standard deviation increase in polygenic risk score was reported, adjusted for age and sex (except for breast cancer). CI confidence interval, OR odds ratio.
PRS were associated with disease status
High polygenic predisposition quantified by PRS was strongly associated with increased risk of developing the corresponding diseases (Figs. 1 and S1). Each standard deviation increase in the PRS was associated with an increased odds of disease, ranging from 1.30-fold (95% CI: 1.18–1.43; p value = 1.4×10−7) for colorectal cancer to 5.25-fold (95% CI: 5.06–5.45; p value < 1.0×10−300) for short stature with narrow 95% CIs (Fig. 1).
Rare and common genetic causes of diseases were largely independent
Between heterozygotes of rare pathogenic variants and nonheterozygotes, we found no distinguishable difference in the distribution of PRS (Fig. S2). Moreover, the effect of rare pathogenic variants and polygenic predisposition appeared to be linearly additive with effect sizes largely unchanged when modeled jointly (Table S4).
Rare pathogenic variants were more common among patients at a low polygenic risk
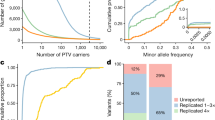
Testing our study hypothesis, we found that among diagnosed cases, the prevalence of rare pathogenic variants was consistently higher among those with a low polygenic predisposition (<50% of the population) than those with a high polygenic predisposition (≥50% of the population). Specifically, the difference in prevalence for each disease was breast cancer (1.55% vs. 0.54%), colorectal cancer (2.44% vs. 2.02%), type 2 diabetes (0.45% vs. 0.15%), osteoporosis (1.34% vs. 0.22%), and short stature (0.70% vs. 0.43%), respectively (Fig. 2a). Stratifying the population by percentile of polygenic predisposition demonstrated large differences in prevalence of rare pathogenic variants (Fig. 2b, c). For example, the prevalence of rare pathogenic variants in individuals in the lowest 10th percentile of the breast cancer PRS was predicted to be >2.13%, compared with <0.42% among women in the highest 10th percentile of the breast cancer PRS (Fig. 2c).

(a) Patients with a low polygenic predisposition (<50% of the population) more likely carried rare pathogenic variants, compared with those with a high polygenic predisposition (≥50% of the population). Dots represent frequencies of diagnosed patients (colored dots) or disease-free individuals (gray dots) carrying corresponding rare pathogenic variants. (b) Rare pathogenic variant heterozygotes more likely had a low polygenic predisposition among diagnosed patients. Each dot represents one diagnosed patient who was a rare pathogenic variant heterozygote. (c) Predicted prevalence of rare pathogenic variants increases among diagnosed patients with a low polygenic predisposition, based on (b). Each standard deviation decrease in the polygenic risk score was associated with 1.89 (95% confidence interval [CI]: 0.98–3.58; p value = 5.4×10−2), 1.32 (95% CI: 0.69–2.62; p value = 0.42), 2.82 (95% CI: 1.14–7.46; p value = 2.9×10−2), 1.80 (95% CI: 0.64–5.17; p value = 0.27), and 1.22-fold (95% CI: 0.86–1.72; p value = 0.26) increased odds of identifying rare pathogenic variants for breast cancer, colorectal cancer, monogenic diabetes, osteoporosis, and short stature if genetic testing were to be performed. Prediction was based on logistic regression.
Rare variants with lower predicted pathogenicity could lessen the association between rare and common genetic causes among patients
For breast cancer, 60 heterozygotes with rare pathogenic variants affecting TP53, PALB2, or ATM were identified, of whom 3 were diagnosed patients. For colorectal cancer, 59 heterozygotes with rare pathogenic variants affecting APC, STK11, BMPR1A, or SMAD4 were identified, of whom 2 were diagnosed patients. Including these additional potentially disease-causing genes led to a slightly weakened association between rare pathogenic variants and disease status (OR = 2.69 [95% CI: 1.39–4.72; p value = 1.4×10−3] for breast cancer and 4.95 [95% CI: 2.52–8.74; p value = 3.6×10−7] for colorectal cancer; Table S5). However, the association between carrying rare pathogenic variants and the PRS remained largely unchanged among diagnosed patients (Table S5).
On the other hand, rare pathogenic variants defined by a relaxed pathogenicity threshold had a largely decreased penetrance (Table S6). For instance, carrying such variants affecting BRCA1 or BRCA2 was only associated with a 1.64-fold (95% CI: 1.07–2.40; p value = 1.7×10−2) increased odds of developing breast cancer. Except for colorectal cancer, the association between rare and common genetic causes substantially lessened among diagnosed patients (Fig. S3). Carrying less pathogenic variants that affected breast cancer, type 2 diabetes, or osteoporosis-related genes was no longer associated with a low polygenic predisposition (Table S6). Notably, carrying these less pathogenic variants in osteoporosis-related genes was not associated with developing osteoporosis (OR = 1.00; 95% CI: 0.51–1.74; p value = 1.00).
Generalizability across different populations
Among a total of 5,358 exome-sequenced nonwhite British individuals, differences in baseline disease prevalence (e.g., breast cancer and diabetes) were evident, despite limited numbers of cases (Table S3). As expected, PRS developed based on the white British population had poor performance when applied to other populations. For example, each standard deviation increase in the PRS for height, the most efficient in the white British population, was associated with merely 1.10-fold (95% CI: 0.95–1.28; p value = 0.20) increased odds of having a short stature among individuals with an African ancestry.
Due to paucity of rare variant heterozygotes and relatively low disease prevalence, the association between carrying rare variants and disease status and that between rare and common genetic predisposition among diagnosed patients were not evaluable for most diseases (Table S7). Nevertheless, we observed suggestive positive correlation between carrying rare pathogenic variants and a low PRS in the African (OR = 3.23 [95% CI: 1.42–7.48; p value = 5.6×10−3] per standard deviation decrease in PRS) and the South Asian (OR = 1.67 [95% CI: 0.77–3.71; p value = 0.20] per standard deviation decrease in PRS) populations with short stature, where we had the largest sample sizes for rare pathogenic variant heterozygotes (Table S7).
DISCUSSION
Identifying rare genetic causes of common disease can help to improve diagnosis and treatment of these diseases. In this study, we found that among diagnosed patients for five common diseases, those with a low polygenic predisposition had a higher likelihood of being heterozygotes of rare pathogenic variants. These findings imply that PRS may assist in prioritizing patients who should undergo sequencing-based genetic tests for known disease-causing genes. Since current costs of PRS generation are substantially lower than sequencing, PRS may help to improve the yield of clinical sequencing studies, especially since a single investment in genome-wide genotyping of approximately US$40 can be used to generate PRS for multiple diseases.
Although rarely emphasized in existing studies, the findings in our study are attributable to a form of “collider bias,”32 where both the rare and common genetic predisposition causally and independently contribute to the disease outcome, becoming a collider. Sample selection or regression analysis conditioning on this collider (among the diagnosed patients) therefore creates a noncausal association between the two genetic risk components. Our findings also point out that this approach may be more helpful in some diseases than others, depending on the strength of the PRS and the genetic architecture of the disease. Specifically, the magnitude of the conditional association may be attenuated when the stratification capacity of the PRS is relatively limited (e.g., the PRS for colorectal cancer in this study), or when the disease of interest has high polygenicity and a single causal variant does not have a strong biological impact (e.g., short stature in this study). It should also be noted that a certain number of rare variant heterozygotes will still be missed if only those categorized into the low–polygenic risk group were to be screened. Therefore, we do not recommend directly applying our proposed principle alone in a clinical context, since we were unable to determine in this study whether a PRS-facilitated decision-making process was advantageous over the traditional decision-making process relying more heavily on family history. In most clinical screening programs that offer a stepwise approach, if a health-care system has sufficient resources (which many currently do not), an individual not having a low PRS could undergo sequencing nonetheless. However, we posit that it will be worth exploring how our proposed approach can be incorporated into current standards to realize its clinical utility.
The high pathogenicity of rare variants is important to our findings. In this study, to ensure high pathogenicity of each identified variant, we mainly focused on rare variants affecting clinically actionable genes or genes showing a dominant inheritance pattern, which presumably have a high penetrance. Consequently, not all potentially pathogenic genes were included in our screening. As demonstrated in sensitivity analyses, including more potentially causal genes increased the number of identified rare variant heterozygotes but those rare variants had lower penetrance. This is not unexpected because the additionally included genes for breast cancer and colorectal cancer mostly have multiple effects, such as the TP53 gene causing Li–Fraumeni syndrome, for which breast cancer is not the only possible outcome.33 Therefore, we believe that a well-calibrated gene panel, e.g., the ClinGen Variant Curation Expert Panel,34 will be required should similar research and clinical applications in other cohorts be attempted.
Our definition of rare pathogenic variants was based on computational prediction rather than clinical standards, since the latter would lead to fewer rare variant heterozygotes being identified due to the rarity of those variants. While our stringent CADD score threshold greatly increased the probability that the identified variants were highly deleterious, other disease-causing variants with a lower predicted pathogenicity were not included. However, as the CADD score decreases, an increasing false positive rate would likely conceal the true associations and could have led to misinterpretation. As we illustrated, including rare variants with lower pathogenicity could substantially weaken the association between rare and common genetic causes among patients. Therefore, we expect a comprehensive curation of rare pathogenic variants in the future as accumulating experimental and clinical evidence will substantially refine our findings. In particular, inclusion of evidently deleterious large chromosomal alterations (e.g., insertion and deletion events) that are extremely rare and underdetected in this study will be necessary.
Our study has several important limitations. First, since the number of exome-sequenced participants in the UK Biobank was not large and this cohort was not established for a specific disease, the number of diagnosed patients for any one trait was limited, compared with larger specifically ascertained cohorts. Consequently, the number of heterozygotes of rare pathogenic variants for each disease was not high. For the same reason, we were not able to define short stature following the clinical standards (>2 standard deviations below the average) as no individual would otherwise carry a rare pathogenic variant. However, it was still promising to observe that associations existed and were consistent for all diseases we investigated, though some estimates showed uncertainty. Finally, with insufficient sample sizes, especially for rare variant heterozygotes and diagnosed patients, of nonwhite British populations in the UK Biobank, our study is not able to robustly investigate the generalizability of our proposed principle across different ethnic groups. Since PRS constructed on European populations generally have worse performance in other populations,35 we might expect the conditional association between rare and common genetic causes to decline. This is partially supported by our results based on short stature in nonwhite British populations. Nevertheless, if a population-specific or a generalizable PRS can be built beyond the UK Biobank, we still anticipate our conclusions to be valid and beneficial in other ancestries.
In summary, the yield of clinical sequencing studies to identify rare pathogenic variants could be increased by stratifying patients with disease by their polygenic risk.
Conclusion
Because the common and rare genetic components both contribute largely to disease pathogenesis and are marginally independent, we propose that rare genetic causes are more prevalent among patients with a low PRS, and that these patients may be prioritized to undergo deep-depth sequencing of the relevant genes.
URLs
Combined Annotation-Dependent Depletion Portal, https://cadd.gs.washington.edu/. UK Biobank, https://www.ukbiobank.ac.uk/.
Data availability
Genome-wide genotyping data, exome-sequencing data, and phenotypic data from the UK Biobank are available upon successful project application.
Code availability
Computer codes used to generate the results in this study are available upon reasonable request to the corresponding author.
References
Visscher PM, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22.
Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017;18:77.
Esplin ED, Oei L, Snyder MP. Personalized sequencing and the future of medicine: discovery, diagnosis and defeat of disease. Pharmacogenomics. 2014;15:1771–1790.
Shepherd M, et al. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med. 2009;26:437–441.
Khera AV, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224.
Inouye M, et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72:1883–1893.
Mars N, et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med. 2020;26:549–557.
Pavan S, et al. Recommendations for choosing the genotyping method and best practices for quality control in crop genome-wide association studies. Front Genet. 2020;11:447.
Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209.
Nagai A, et al. Overview of the BioBank Japan Project: study design and profile. J Epidemiol. 2017;27:S2–S8.
Chen Z, et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol. 2011;40:1652–1666.
Fry A, et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–1034.
Yengo L, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–3649.
Ranke MB. Towards a consensus on the definition of idiopathic short stature. Horm Res. 1996;45:64–66.
Kalia SS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255.
Misra S, Owen KR. Genetics of monogenic diabetes: present clinical challenges. Curr Diab Rep. 2018;18:141.
Makitie RE, et al. New insights into monogenic causes of osteoporosis. Front Endocrinol (Lausanne). 2019;10:70.
Rappold G, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007;44:306–313.
Grunauer M, Jorge AAL. Genetic short stature. Growth Horm IGF Res. 2018;38:29–33.
Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Rentzsch P, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894.
Mavaddat N, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet. 2019;104:21–34.
Njeh CF, et al. Assessment of bone status using speed of sound at multiple anatomical sites. Ultrasound Med Biol. 2001;27:1337–1345.
Forgetta V, et al. Development of a polygenic risk score to improve screening for fracture risk: a genetic risk prediction study. PLoS Med. 2020;17:e1003152.
Wood AR, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186.
Vilhjalmsson BJ, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–592.
Weigl K, et al. Genetic risk score is associated with prevalence of advanced neoplasms in a colorectal cancer screening population. Gastroenterology. 2018;155:88–98 e10.
Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9:e1003348.
Antoniou AC, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506.
Southey MC, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. 2016;53:800–811.
Easton DF, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257.
Greenland S, Pearl J, Robins JM. Causal diagrams for epidemiologic research. Epidemiology. 1999;10:37–48.
Malkin D, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238.
Rivera-Munoz EA, et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat. 2018;39:1614–1622.
Martin AR, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–591.
Acknowledgements
This research has been conducted using the UK Biobank Resource under application number 27449. J.B.R.’s research group is supported by the Canadian Institutes of Health Research, the Lady Davis Institute of the Jewish General Hospital, the Canadian Foundation of Innovation, and the Fonds de Recherche Québec Santé (FRQS). T.L. is supported by an FRQS Doctoral Training Fellowship and a McGill University Faculty of Medicine Scholarship. J.B.R. is supported by an FRQS Clinical Research Scholarship. These funding agencies had no role in the design, implementation, or interpretation of this study. This study was enabled in part by support provided by Calcul Québec and Compute Canada.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lu, T., Zhou, S., Wu, H. et al. Individuals with common diseases but with a low polygenic risk score could be prioritized for rare variant screening. Genet Med 23, 508–515 (2021). https://doi.org/10.1038/s41436-020-01007-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01007-7
Keywords
This article is cited by
-
Statistical methods for assessing the effects of de novo variants on birth defects
Human Genomics (2024)
-
Lung cancer in patients who have never smoked — an emerging disease
Nature Reviews Clinical Oncology (2024)
-
Genetic Evaluation for Monogenic Disorders of Low Bone Mass and Increased Bone Fragility: What Clinicians Need to Know
Current Osteoporosis Reports (2024)
-
Non-BRCA1/BRCA2 high-risk familial breast cancers are not associated with a high prevalence of BRCAness
Breast Cancer Research (2023)
-
From target discovery to clinical drug development with human genetics
Nature (2023)
