
Abstract
Purpose
Mitochondrial DNA (mtDNA) depletion syndrome (MDDS) encompasses a group of genetic disorders of mtDNA maintenance. Mutation of RRM2B is an uncommon cause of infantile-onset encephalomyopathic MDDS. Here we describe the natural history of this disease.
Methods
Multinational series of new genetically confirmed cases from six pediatric centers.
Results
Nine new cases of infantile-onset RRM2B deficiency, and 22 previously published cases comprised a total cohort of 31 patients. Infants presented at a mean of 1.95 months with truncal hypotonia, generalized weakness, and faltering growth. Seizures evolved in 39% at a mean of 3.1 months. Non-neurological manifestations included respiratory distress/failure (58%), renal tubulopathy (55%), sensorineural hearing loss (36%), gastrointestinal disturbance (32%), eye abnormalities (13%), and anemia (13%). Laboratory features included elevated lactate (blood, cerebrospinal fluid (CSF), urine, magnetic resonance (MR), spectroscopy), ragged-red and cytochrome c oxidase–deficient fibers, lipid myopathy, and multiple oxidative phosphorylation enzyme deficiencies in skeletal muscle. Eight new RRM2B variants were identified. Patients with biallelic truncating variants had the worst survival. Overall survival was 29% at 6 months and 16% at 1 year.
Conclusions
Infantile-onset MDDS due to RRM2B deficiency is a severe disorder with characteristic clinical features and extremely poor prognosis. Presently management is supportive as there is no effective treatment. Novel treatments are urgently needed.
Similar content being viewed by others


INTRODUCTION
RRM2B (OMIM 604712) is a nuclear gene encoding the small p53 inducible subunit (p53R2) of the ribonucleotide reductase (RNR) complex. RNR is a heterotetramer1,2,3 composed of two R1 subunits (encoded by RRM1), and two R2 (encoded by RRM2) or p53R2 (encoded by RRM2B) subunits. RNR localizes to the cytosol4 and reduces ribonucleotide diphosphates to deoxyribonucleotide diphosphates, which is important in keeping the mitochondrial deoxyribonucleotide triphosphate (dNTP) pool replete. These dNTPs are then thought to be relocated to the nucleus and mitochondria for DNA synthesis.
Unlike nuclear DNA, which replicates during the S phase of the cell cycle before mitosis, mitochondrial DNA (mtDNA) is synthesized throughout the nonproliferative phase (G0) and thus requires a steady pool of dNTPs. Therefore, RNR exists in two forms: that which supports de novo dNTP synthesis during the S phase (composed of 2R1, 2R2 subunits) and that which is responsible for maintenance of dNTP pools in G0 (composed of 2R1, 2p53R2). p53 is a tumor suppressor gene that is activated during DNA damage. p53R2 is thought to have two roles: provision of dNTPs for DNA repair following nuclear DNA damage2 and also a “housekeeping role” in maintaining dNTP pools needed for mtDNA replication throughout the cell cycle in both dividing and nondividing cells.5,6
Mitochondrial DNA depletion syndrome (MDDS) encompasses a group of nuclear gene disorders affecting key enzymes involved in the synthesis of mtDNA and its replication. This includes several genes involved in either mtDNA replication itself or in the synthesis of nucleosides that are incorporated into mtDNA at the replication fork.7,8 MDDS is classically categorized into different phenotypic subtypes, each with an expanding number of causative disease genes. These phenotypes are myopathic (caused by mutations in TK2, SLC25A4, MGME1), encephalomyopathic (RRM2B, FBXL4, ABAT), hepatocerebral (TFAM, MPV17, DGUOK, SUCLA2, SUCLG1), Alpers syndrome (POLG, TWNK), and mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) (TYMP, rarely POLG and RRM2B).6,7,8,9 Mutations of RRM2B may be associated with several different phenotypes, including infantile-onset MDDS caused by autosomal recessive inheritance of biallelic pathogenic variants.3,5,10,11,12,13,14,15,16 Other phenotypes include multiple mtDNA deletions in children and adults that are caused by either autosomal recessive inheritance of biallelic pathogenic variants or autosomal dominant inheritance of a single pathogenic variant,17,18,19,20,21 Kearns–Sayre syndrome,19 MNGIE,22 and, most recently, acute liver failure.23
We describe nine new cases of infantile-onset encephalomyopathic RRM2B MDDS, reporting their detailed phenotype, investigations, treatment, and outcome. Taken together with a further 22 published cases, we describe the natural history of 31 patients with infantile RRM2B deficiency.
MATERIALS AND METHODS
Patient cohort
Nine new patients were recruited from three centers in the UK, two centers in the United States, and one center in France. Patients were included if they had confirmed biallelic RRM2B variants and an infantile-onset clinical phenotype, and were not previously published. Data were collected retrospectively by clinicians in each center using a standardized questionnaire. This study was approved by Great Ormond Street Hospital (GOSH) as a clinical audit.
Molecular studies in new patients
Mitochondrial DNA copy number relative to nuclear DNA was estimated and compared with age-matched normal controls by real-time quantitative polymerase chain reaction (PCR).24 For the UK cases, this was performed as described previously except that the nuclear DNA probe was labeled with Vic and the assays were performed simultaneously using a PE7500 real-time PCR instrument (Applied Biosystems, Foster City, CA, USA). All exons and exon–intron boundaries of RRM2B (GenBank accession number NM_015713.4, exons 1–9 numbered sequentially) were Sanger sequenced using capillary electrophoresis in a 3730 DNA Analyzer (Applied Biosystems). For probands in whom Sanger sequencing only identified a single heterozygous pathogenic variant, RRM2B exon copy number was assessed by multiplex ligation-dependent probe amplification (SALSA MLPA P089-B1, MRC-Holland, Amsterdam, The Netherlands). For two cases, genetic diagnoses were obtained by genome sequencing in a rapid genome sequencing project for critically ill infants.25 For a further case, the causal RRM2B variants were identified by exome sequencing as previously described.26
Literature search
We collected data from all published cases of infantile-onset RRM2B-related MDDS by searching PubMed for relevant publications. Search terms were “RRM2B” OR “ribonucleotide reductase” OR “p53R2” with filters for English language publications and human species (search completed 21 October 2018). This yielded 1948 publications including 10 papers containing relevant cases.
Statistical analyses
Stata software version 15 was used for all statistical analyses (https://www.stata.com/). A P value < 0.05 was considered statistically significant.
Modeling
FoldX server was used to model the new missense variants.27
RESULTS
Patient cohort
Nine previously unreported cases are included in our multinational cohort (patients A–I). They presented with symptoms at an average of 2.3 months (range 0–4 months). Twenty-two further cases were identified from the literature (patients 1–22), making a total of 31 cases of infantile-onset RRM2B-deficient MDDS.3,5,10,11,12,13,14,15,16
Molecular analyses
Mitochondrial DNA studies
Residual mtDNA levels in skeletal muscle in the new cohort of nine previously unreported infants with biallelic RRM2B variants ranged from 2% to 20% (median 4.5%), which is far below the accepted diagnostic limit of 30% for MDDS.28
Novel pathogenic RRM2B variants
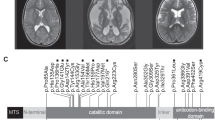
We identified eight novel pathogenic RRM2B variants not present in the gnomAD database (https://gnomad.broadinstitute.org/gene/ENSG00000048392). These are c.599G>A p.Gly200Glu, c.313G>A p.Glu105Lys, c.181G>C p.Ala61Pro, c.165G>A p.Met55Ile, c.(321+1_322–1)_(684+1_685-1)del (exon 4–6 deletion), c.635_636insAAG p.Gly212_Leu213insSer, c.400C>G p.His134Asp, and c.59C>G p.Ser20*. The six missense and insertion variants are found in amino acid positions with a high degree of evolutionary conservation across species (Clustal Omega [http://www.clustal.org/]; see Supplementary Fig. 1 for alignments, Fig. 1a and Supplementary Fig. 2 for the biallelic variants identified in the cohort); at these positions, the amino acids of the human sequence are also shared by 65–96% of RRM2B orthologs (n = 150 orthologs; data from Consurf).

Novel pathogenic RRM2B variants in new patient cohort. (a) The nine exons (E1–9) of RRM2B messenger RNA (mRNA) showing the location of each of the variants identified in the nine new patients (blue for new variants and green for previously reported variants). p53R2 comprises 351 amino acid residues. Each exon is labeled with the segment of the amino acid chain it encodes. Variants in the 22 previously reported patients are indicated in black. fs frameshift. Asterisk (*) denotes stop codon. (b) 3D structure of a RRM2B dimer depicting the location of new nontruncating variants observed in the new cases. The two subunits of the RRM2B dimer, taken from the reported structure (Protein Data Bank [PDB] code 3HF1), are shown in ribbon representation. The wild-type amino acids bearing the novel variants from this study are shown as sticks, colored yellow (for missense changes) and cyan (for indel). The di-iron metal center for one RRM2B subunit is represented by the two Fe atoms shown as spheres.
The six nontruncation variants are distributed across the N-terminal two-thirds of the RRM2B protein, a 351-residue polypeptide (Fig. 1b). The crystal structure of human RRM2B reveals a homodimer with an active site di-iron cofactor from each monomer.1 Two variants, p.Ala61Pro and p.Gly200Glu, result in marked changes to the amino acid side-chains, and are predicted by the FoldX server to reduce protein stability (ΔΔG 3.97 and 11.60 kcal/mol respectively). His134 is one of the iron-ligating residues in the structure, hence the p.His134Asp variant likely interferes with metal binding, and is also predicted by FoldX to reduce protein stability (ΔΔG 6.50 kcal/mol). Glu105 is located at the dimer interface, forming salt bridges with Arg40 and Arg121 from the opposite dimer subunit. The p.Glu105Lys variant could therefore interfere with structural integrity of the homodimer. The p.Met55Ile and p.Gly212_Leu213insSer variants are located at the protein exterior, away from the core active centers; the effects of these substitutions are less clear from the structure.
Disease course of nine new cases
The disease phenotype was multisystemic, with an initial myopathic presentation followed by evolution of respiratory distress and failure leading to assisted ventilation in all cases. Multisystem involvement included the neurological, renal, cardiac, respiratory, gastrointestinal (GI), and hematological systems, and the eyes, hearing, and growth. Table 1 and Fig. 2 detail the biochemical and histological findings. Supplementary Fig. 3 illustrates the neurological, respiratory, renal, and GI features and growth of the nine new cases. Supplementary Fig. 4A–C illustrates the treatments administered to the new cases, including respiratory support, supportive feeding, and cofactor supplementation. Supplementary Fig. 4D illustrates the survival outcomes for the new cases. By 6 months 8/9 patients had already developed respiratory failure and 6/9 had died. All nine patients died by 22 months. The causes of death are illustrated in Supplementary Fig. 4E.

Histopathological findings associated with RRM2B deficiency. The images show the histochemical and ultrastructural features of patients C, D, G, and I. On hematoxylin and eosin (HE) staining (a–d), there were myopathic features, increased variation in fiber size with excess small fibers, and vacuoles. On Gömöri trichrome (GT) staining (e–h), there was accumulation of mitochondria in the vacuolated fibers and there were some ragged-red fibers. Lipid staining with oil red O (i–l) showed marked excess lipid in the vacuolated fibers. Succinate dehydrogenase (SDH) histochemistry showed fibers with increased staining (m–p). Cytochrome c oxidase showed frequent negative fibers in all four patients (q–t). Electron microscopy was available in patients D, G, and I and showed clusters of enlarged atypical mitochondria (u–w). Nemaline rods were also present (not illustrated) in patients G and I. Patient D had two muscle biopsies 4 months apart, the second taken after a low-fat diet. The two biopsies were similar except for reduced lipid following the low-fat diet. All the images of this patient show the first biopsy except for (n), which is from the second. The table summarizes muscle biopsy findings for 8/9 new patients for whom this was available. CoQ coenzyme Q, COX cytochrome c oxidase, EM electron microscopy, NADH reduced nicotinamide adenine dinucleotide, ND not done, RBF ragged-blue fibers, RRF ragged-red fiber. Scale Bars: a–t: 50 µm, u–w: 2 µm.
Natural history of cohort of 31 patients
Considering the entire cohort of 31 infants with RRM2B deficiency, median age of symptom onset was 2 months (mean 1.95 months, range 0–6 months). The most frequent presenting features were muscular hypotonia, feeding difficulty, failure to thrive, hearing loss, and respiratory distress. The clinical phenotype was multisystemic. Neurological involvement occurred in all cases: encephalopathy in 15/31 (48%), seizures in 12/31 (39%) presenting at an average age of 3.1 months (range 20 days–7 months), respiratory involvement in 18/31 (58%), renal involvement in 17/31 (55%), growth abnormalities in 16/31 (51%), hearing loss in 11/31 (36%), GI involvement in 10/31 (32%), eye abnormalities in 4/31 (13%), and anemia in 4/31 (13%) of cases. The most consistent biochemical abnormality was elevated lactate seen in blood, cerebrospinal fluid (CSF), and urine, followed by elevated alanine and mildly elevated creatine kinase. The most frequent skeletal muscle histopathological features were COX-deficient fibers, abnormal lipid deposition, and ragged-red fibers. The most frequent oxidative phosphorylation (OXPHOS) biochemical abnormalities in muscle were decreased activities of complexes I, III, and IV with compensatory increases in both complex II and citrate synthase activities. Neuroimaging most frequently showed a structurally normal brain, and less frequently evidence of demyelination, white matter abnormality, or generalized atrophy (Table 2). Elevated lactate peak was frequently observed on magnetic resonance (MR) spectroscopy. Detailed clinical, biochemical, and radiological features of all 31 cases are summarized in Table 2.
Survival outcomes for the cohort were poor. A Kaplan–Meier survival curve of all 31 cases of RRM2B-associated infantile MDDS (Fig. 3) shows that survival at 3 months was 64.5% (20/31), at 6 months 29% (9/31), at 1 year 16% (5/31), and at 2 years 6% (2/31).

Kaplan–Meier curve of survival of the 31-patient cohort of RRM2B-associated mitochondrial DNA depletion syndrome (MDDS). The figure shows survival of 31 cases including 9 new cases from this cohort and 22 previously published cases. Three-month survival was 64.5%, six-month survival was 29%, one-year survival was 16%, two-year survival was 9.6%.
Phenotypic correlations
Mitochondrial DNA content
Patients with lower residual mtDNA levels in skeletal muscle tended to die earlier, however this observation did not meet statistical significance (Spearman rank test, p = 0.06). There was no statistically significant correlation between residual mtDNA content and age of onset (Spearman rank test, p = 0.44) or number of organ systems affected (Spearman rank test, p = 0.14). We compared patients who developed seizures (12/31) as a subgroup with patients who did not develop seizures (19/31) and noted no difference in average mtDNA levels between these two groups (Mann–Whitney U test, p = 0.85). Similarly, we noted no significant difference in average residual mtDNA levels in patients who developed renal disease (n = 17) compared with those who did not develop renal disease (n = 14) (Mann–Whitney U test, p = 0.29).
Age of onset of symptoms
Infants presenting earlier appeared to die earlier, however, this did not reach statistical significance (Spearman rank test, p = 0.07). There was no correlation between age of onset and number of organs affected (Spearman rank test, p = 0.20, nor between age of onset and development of seizures (Mann–Whitney U test, p = 0.88). Interestingly patients who developed renal disease presented earlier (median age of onset for patients who developed renal disease was 2 months versus 3 months for those without renal disease, Mann–Whitney U test, p = 0.01*).
Age of death
There was no correlation between age of death and number of organs affected (Spearman rank test, p = 0.42), nor between age of death and occurrence of seizures (Mann–Whitney U test, p = 0.56). Patients with renal disease tended to die earlier, however this did not reach statistical significance (Mann–Whitney U test, p = 0.07).
Genotype–phenotype correlations
Patients were divided into two groups based on genotype. Group 1 included seven patients whose pathogenic variants are expected to result in the formation of only truncated messenger RNA (mRNA), which is likely to be subject to nonsense-mediated decay (five with biallelic nonsense variants [patients 1–3, 21, 22], one with biallelic frameshift variants [patient 19], and one compound heterozygote with a nonsense variant and a splice site variant [patient I]). Group 2 patients included the remaining 24 patients whose variants are not expected to result in the formation of solely truncated mRNA (15 patients with biallelic missense variants [patients A, B, D, G, H, 6, 8, 11, 13–18, 20], 3 compound heterozygotes with a missense variant and a splice site variant [patients 4, 5, 12], 3 compound heterozygotes with a missense variant and a truncating variant [patients C, 9, 10], 2 patients with biallelic in-frame insertions [patients E, F], and 1 compound heterozygote for a missense variant and an in-frame deletion [patient 7]).
Group 1 patients had a worse prognosis than group 2. Median age of death was 2.5 months for group 1 compared with 5 months for group 2 (Mann–Whitney U test, p = 0.002**). There was no statistically significant difference between age of onset or number of organs affected between the two groups (Mann–Whitney U test, p = 0.33 and 0.16 respectively), nor any correlation between genotypic group and development of seizures (Chi squared test, p = 0.53) or renal disease (Chi squared test, p = 0.31).
DISCUSSION
Here we have shown that infantile MDDS due to biallelic RRM2B variants presents in the very first months of life with hypotonia, poor feeding, and faltering growth leading to hospitalization. Elevated blood and CSF lactate may be the clue to an underlying mitochondrial disease, prompting multisystem assessment revealing involvement of other organs, frequently including sensorineural hearing loss (SNHL) and renal tubulopathy/nephrocalcinosis. Infants may also develop GI symptoms such as recurrent vomiting/reflux. All deteriorate with respiratory insufficiency/failure leading to assisted ventilation. Generalized seizures may develop later in the disease course. This infantile clinical phenotype is distinct from pediatric and adult-onset disease associated with multiple mtDNA deletions due to recessive (biallelic) and dominant (heterozygous) RRM2B variants, where progressive external ophthalmoplegia (PEO), ptosis, and proximal muscle weakness are the most frequent features and encephalopathy, GI, and renal involvement are uncommon.21 Interestingly, SNHL is present to the same degree (36% of infantile-onset MDDS and 36% of cases with pediatric/adult-onset disease).
Although we have shown that the phenotype of infantile MDDS due to RRM2B deficiency is relatively homogeneous, we did observe some phenotypic heterogeneity within the cohort. Notably, although MDDS due to RRM2B deficiency is classically described as encephalomyopathic,6 encephalopathy was seen in 15/31 cases (49%) and seizures were observed in only 12 infants (39%). It is possible that some patients died due to severe myopathy before the development of encephalopathy or indeed that they would never have developed encephalopathy. Incomplete penetrance of neurological features is seen in other genetic causes of MDDS, e.g., infantile-onset DGUOK deficiency where individuals may develop liver disease, which can be fatal, or go on to develop multisystemic disease with additional brain involvement including encephalopathy. Conversely in our cohort, renal tubulopathy was seen in 17/31 (55%) of cases, and SNHL in 12/31 (39%). The high incidence of renal tubulopathy in infants with MDDS is reminiscent of the phenotype of the rrm2b−/− exon 3 and 4 deletion mouse model in which renal failure is the most prominent feature and cause of death.29
Mutational analysis of the new patients in our cohort revealed that several variants seen in patients with infantile MDDS have also been seen in children and adults with multiple mtDNA deletions presenting with PEO and myopathy. This confirms previous reports suggesting that missense variants may behave recessively (homozygous or compound heterozygous) as well as in a heterozygous dominant negative manner in different individuals. The presence of normal p53R2 protein in cases of heterozygosity may not always rescue the presence of mutated p53R2, owing to a dominant negative effect that may be mediated by disruption of normal allosteric interactions between the heterotetrameric subunits of the RNR complex.21
In this study we sought to determine whether there are any phenotypic and genotype–phenotype correlations in RRM2B-associated MDDS. This is important for prognostication for affected infants and in future may enable assessment of suitability for candidate therapies. As MDDS is defined as residual mtDNA <30% of age-matched healthy controls, all of the patients met this criterion.28 In this natural history study, we investigated whether mtDNA quantification can be used not just in diagnosis but also in predicting severity of disease. As it was not possible to apply a specific scoring system, for example the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS), in entirety to each patient, the markers of severity of disease that we analyzed in this study were age of onset, presence of seizures, number of organs affected, and survival. Taken pairwise, no statistically significant correlation was seen between these markers of disease severity and mtDNA quantification. Nevertheless, important trends were revealed: earlier disease onset tended to be associated with presence of renal disease later in the disease course and an earlier mortality. While there was no statistical correlation between the presence of seizures and age of death, for the 12 patients in the cohort who did develop seizures, death followed after an average of 2.9 months (median 1.2 months, range 0–15 months) after the appearance of seizures, defining a point of significant clinical deterioration in the disease course. Therefore, we suggest that the presence of seizures indicates significant brain involvement and therefore severe disease.
When we grouped patients into two variant subgroups we found that patients with biallelic variants resulting in truncated transcripts had a relatively shorter survival compared with patients with missense/in-frame insertion/deletions resulting in formation of mRNA transcripts that are aberrant but not subject to degradation by cellular nucleases. This finding was statistically significant despite a small cohort and is consistent with our findings for other natural history studies in mitochondrial disease.30
It should be noted that there is a limitation of using age of death as a marker of disease severity since in some cases death followed redirection of care after recognizing futility of treatment. Severe disease may paradoxically be associated with a longer than expected lifespan, owing to prolonged artificial ventilation. Another consideration is that meaningful correlation between percentage mtDNA levels and genotype based on protein truncation could not be determined due to small numbers. Finally, it is assumed that the formation of truncated mRNA will result in mRNA degradation and therefore little enzyme activity. However, an exception to this has been demonstrated in myocytes of adult patients with exon 9 deletions where mRNAs escaped nonsense-mediated decay and western blots showed persistent presence of truncated protein.17
We have demonstrated that patients with biallelic variants resulting in truncated mRNA transcripts have the poorest prognosis overall. This is an important finding in terms of prognostication and appears to be the most robust genotype–phenotype correlation. The prognosis overall however is extremely poor, with a six-month survival of 29% and a one-year survival of 16%, underscoring the need to find new effective treatments.
In RRM2B deficiency, inability to reduce ribonucleotide diphosphates to deoxyribonucleotide diphosphates (dNDPs) results in mtDNA depletion. For a different form of myopathic MDDS, thymidine kinase 2 (TK2) deficiency, it has been proposed that bypassing the defective step of nucleoside salvage by supplementation with deoxyribonucleotides may represent a therapeutic strategy for MDDS. This has been trialed in tk2-deficient mice with some success. A combination of orally supplemented deoxycytidine monophosphate (dCMP) and deoxythymidine monophosphate (dTMP) delayed disease onset and prolonged survival but did not prevent death in a knock-in model of tk2 deficiency.31 Deoxyribonucleotide monophosphates appeared able to cross the immature blood–brain barrier before P13 but further development of the blood–brain barrier after P13 was likely to account for central nervous system (CNS) disease at P29 and ultimate death despite therapy. Similar results were seen with oral supplementation with deoxycytidine (dC) and deoxythymidine (dT) in mice with tk2 deficiency, which is thought to be due to compensatory upregulation of the cytosolic enzymes tk1 and dck.32 For RRM2B deficiency, in vivo studies using a suitable animal knockout model are yet to be undertaken, since the existing animal model does not effectively recapitulate human disease.29
For RRM2B deficiency, in vitro studies of supplementation with deoxynucleosides in cultured human cells yielded contradictory results. RRM2B-deficient patient fibroblasts do not express mtDNA depletion in culture unless they are exposed to ethidium bromide or prolonged serum starvation.33 Deoxynucleoside supplementation increased intracellular dNTP pools and normalized mtDNA synthesis in ethidium bromide–treated quiescent patient fibroblasts harboring a lethal homozygous missense RRM2B variant.34 However, in a second study, supplementation with deoxynucleotide monophosphates (dNMPs) in cultured myotubules/myoblasts from another patient was ineffective in restoring mtDNA copy number.35 In vivo delivery of all four deoxyribonucleosides via any route has not been investigated in any model of MDDS. Furthermore, the CNS penetrance of systemically delivered deoxyribonucleosides in humans is unknown. The half-life of adenosine is extremely brief when delivered intravenously and there is a risk of toxicity since adenosine induces atrioventricular node blockade. It is clear that in the presence of many unknowns, detailed pharmacodynamic, pharmacokinetic, and efficacy studies in a knockout animal model of RRM2B deficiency that effectively recapitulates neurological disease seen in humans will be essential to answer these questions.
Prior to designing a clinical trial for an experimental treatment for RRM2B-related infantile MDDS in humans, it will be necessary to determine at which time point CNS involvement is considered irreversible. This should consider both the natural history of disease in humans and the optimal timepoint for effective drug delivery in preclinical drug trials using an appropriate animal model. Difficult decisions will need to be made in the advent of experimental therapies as to whether a specific patient is deemed to have reversible disease, especially since conventional neuroimaging is often normal early on in infants who have quite severe functional neurodisability, and seizures are not universally seen. However, in cases for which there are structural abnormalities on neuroimaging or prolonged/refractory electrical or clinical seizures, we suggest that CNS disease has progressed beyond the point of reversibility.
Whether or not irreversible disease should be treated with an experimental treatment ought to be considered on a case-by-case basis utilizing existing ethical frameworks. This is predicated on determining whether the disease-modifying treatment is likely to improve the quality of life rather than just prolonging life. The Royal College of Paediatrics and Child Health have provided an ethical framework that states that the best interests of the child need to be considered to “determine whether or not there is an overall benefit in prolonging life because of the adverse impact entailed.”36 The ethical implications of prolonging life with long-term ventilation in the context of commencement of an experimental therapy need to be weighed carefully against those of redirection of supportive care. Indeed, long-term ventilation may be requested as a bridge to emerging or experimental therapies and its use in this setting is difficult to evaluate.37 An ethics committee should be called upon to meet to assess the individual clinical case considering the opinion of the infant’s parents and available second opinions provided by other clinical teams.
It is clear that the natural history of infantile-onset RRM2B MDDS is characterized by rapid neurological disease progression. Prompt diagnosis is essential for optimal management of these infants, particularly avoidance of prolonged invasive ventilation as currently there are no effective disease-modifying therapies for this and most other mitochondrial diseases.38 In the future, if a clinical trial of a disease-modifying therapy is to be undertaken, clinical and ethical discussions regarding suitability for trial recruitment of the affected infant need to commence soon after a genetic diagnosis is made.
To facilitate more rapid diagnosis, we suggest that where there is a high index of clinical suspicion indicated by a combination of several of the following six features: hypotonia developing insidiously after birth, renal tubulopathy, sensorineural hearing loss, feeding difficulty, muscle weakness, and respiratory failure, targeted single-gene sequencing for RRM2B should be prioritized over muscle biopsy and exome sequencing. Where the index of clinical suspicion is lower, that is, when only a few of these features are present, the differential diagnosis includes potentially treatable disorders such as spinal muscular atrophy,12 benign reversible infantile respiratory chain deficiency disorders,39 congenital myopathies, and other causes of MDDS, e.g., TK2 deficiency. In this scenario a rapid exome sequencing approach with consideration of early muscle biopsy is more appropriate. Of note, creatine kinase levels are normal or only mildly elevated in RRM2B deficiency effectively distinguishing this disorder from myopathic MDDS due to TK2 deficiency.40
Conclusions
Infantile-onset RRM2B-related MDDS should be suspected in cases where mtDNA depletion has been demonstrated in an infant with a combination of encephalomyopathy, respiratory failure, and renal or hearing impairment. The majority of infants are already symptomatic with hypotonia and faltering growth well before presentation with respiratory failure to emergency medical services. The disease evolves relentlessly with a one-year survival of 16%. Biallelic truncating variants predict the worst survival outcome. While awaiting a genetic diagnosis there is evolving brain injury, implying a narrow therapeutic window for treatment with disease-modifying therapies, should these become available. The usefulness of deoxyribonucleoside supplementation remains to be elucidated in an animal model that adequately recapitulates human neurological disease before considering clinical trials in humans.
References
Smith P, Zhou B, Ho N, et al. 2.6 A X-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry. 2009;48:11134–11141.
Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49.
Bourdon A, Minai L, Serre V, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780.
Pontarin G, Fijolek A, Pizzo P, et al. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc Natl Acad Sci USA. 2008;105:17801–17806.
Kollberg G, Darin N, Benan K, et al. A novel homozygous RRM2B missense mutation in association with severe mtDNA depletion. Neuromuscul Disord. 2009;19:147–150.
El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10:186–198.
Viscomi C, Zeviani M. MtDNA-maintenance defects: syndromes and genes. J Inherit Metab Dis. 2017;40:587–599.
Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15:40–52.
Gorman GS, Taylor RW. RRM2B-related mitochondrial disease. In: Adam MP, Ardinger HH, Pagon RA, (eds.) GeneReviews. Seattle, WA: University of Washington; 1993.
Bornstein B, Area E, Flanigan KM, et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord. 2008;18:453–459.
Acham-Roschitz B, Plecko B, Lindbichler F, et al. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–304.
Spinazzola A, Invernizzi F, Carrara F, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009;32:143–158.
Stojanovic V, Mayr JA, Sperl W, Barisic N, Doronjski A, Milak G. Infantile peripheral neuropathy, deafness, and proximal tubulopathy associated with a novel mutation of the RRM2B gene: case study. Croat Med J. 2013;54:579–584.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
McCormack SE, Gai X, Place E, Falk MJ. Mitochondrial DNA depletion syndromes presenting in childhood. In: Saneto RP, Parikh S, Cohen BH, (eds.) Mitochondrial case studies. London: Academic Press; 2016. p. 187–198.
Kropach N, Shkalim-Zemer V, Orenstein N, Scheuerman O, Straussberg R. Novel RRM2B mutation and severe mitochondrial DNA depletion: report of 2 cases and review of the literature. Neuropediatrics. 2017;48:456–462.
Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, Suomalainen A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet. 2009;85:290–295.
Fratter C, Raman P, Alston CL, et al. RRM2B mutations are frequent in familial PEO with multiple mtDNA deletions. Neurology. 2011;76:2032–2034.
Pitceathly RD, Fassone E, Taanman JW, et al. Kearns–Sayre syndrome caused by defective R1/p53R2 assembly. J Med Genet. 2011;48:610–617.
Takata A, Kato M, Nakamura M, et al. Exome sequencing identifies a novel missense variant in RRM2B associated with autosomal recessive progressive external ophthalmoplegia. Genome Biol. 2011;12:R92.
Pitceathly RD, Smith C, Fratter C, et al. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain. 2012;135(pt 11):3392–3403.
Shaibani A, Shchelochkov OA, Zhang S, et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. 2009;66:1028–1032.
Valencia CA, Wang X, Wang J, et al. Deep sequencing reveals novel genetic variants in children with acute liver failure and tissue evidence of impaired energy metabolism. PLoS ONE. 2016;11:e0156738.
Poulton J, Sewry C, Potter CG, et al. Variation in mitochondrial DNA levels in muscle from normal controls. Is depletion of mtDNA in patients with mitochondrial myopathy a distinct clinical syndrome. J Inherit Metab Dis. 1995;18:4–20.
Mestek-Boukhibar L, Clement E, Jones WD, et al. Rapid paediatric sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. J Med Genet. 2018;55:721–728.
Nicholls TJ, Nadalutti CA, Motori E, et al. Topoisomerase 3ɑ is required for decatenation and segregation of human mtDNA. Mol Cell. 2018;69:9–23 e26.
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388.
Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child. 2009;94:3–5.
Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat Genet. 2003;34:440–445.
Keshavan N, Rahman S. Natural history of mitochondrial disorders: a systematic review. Essays Biochem. 2018;62:423–442.
Garone C, Garcia-Diaz B, Emmanuele V, et al. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med. 2014;6:1016–1027.
Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, et al. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol. 2017;81:641–652.
Pontarin G, Ferraro P, Rampazzo C, et al. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J Biol Chem. 2011;286:11132–11140.
Pontarin G, Ferraro P, Reichard P, Bianchi V. Out of S-phase: shift of subunits for ribonucleotide reduction. Cell Cycle. 2012;11:4099–4100.
Bulst S, Holinski-Feder E, Payne B, et al. In vitro supplementation with deoxynucleoside monophosphates rescues mitochondrial DNA depletion. Mol Genet Metab. 2012;107:95–103.
Larcher V, Craig F, Bhogal K, et al. Making decisions to limit treatment in life-limiting and life-threatening conditions in children: a framework for practice. Arch Dis Child. 2015;100(suppl 2):s3–s23.
Ray S, Brierley J, Bush A, et al. Towards developing an ethical framework for decision making in long-term ventilation in children. Arch Dis Child. 2018;103:1080–1084.
Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560–2574.
Horvath R, Kemp JP, Tuppen HA, et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain. 2009;132(pt 11):3165–3174.
Garone C, Taylor RW, Nascimento A, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. 2018;55:515–521.
Acknowledgements
S.R. is supported by a Great Ormond Street Hospital Children’s Charity Research Leadership Award (V1260), the National Institute for Health Research (NIHR) Great Ormond Street Hospital Biomedical Research Center, and the Lily Foundation. R.W.T. is supported by the Wellcome Center for Mitochondrial Research (203105/Z/16/Z), the Medical Research Council (MRC) Center for Translational Research in Neuromuscular Disease, Mitochondrial Disease Patient Cohort (UK) (G0800674), the Lily Foundation, the UK NIHR Biomedical Research Center for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust, the MRC/Engineering and Physical Sciences Research Council (EPSRC) Molecular Pathology Node, and the UK NHS Highly Specialised Service for Rare Mitochondrial Disorders of Adults and Children. E.W.S. was funded by an Medical Research Council (MRC) PhD studentship. The views expressed are those of the authors(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Keshavan, N., Abdenur, J., Anderson, G. et al. The natural history of infantile mitochondrial DNA depletion syndrome due to RRM2B deficiency. Genet Med 22, 199–209 (2020). https://doi.org/10.1038/s41436-019-0613-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0613-z
