
Abstract
Purpose
To date, heterozygous or homozygous COL12A1 variants have been reported in 13 patients presenting with a clinical phenotype overlapping with collagen VI–related myopathies and Ehlers–Danlos syndrome (EDS). The small number of reported patients limits thorough investigation of this newly identified syndrome, currently coined as myopathic EDS.
Methods
DNA from 78 genetically unresolved patients fulfilling the clinical criteria for myopathic EDS was sequenced using a next-generation panel of COL12A1, COL6A1, COL6A2, and COL6A3.
Results
Among this cohort, we identified four pathogenic heterozygous in-frame exon skipping (∆) defects in COL12A1, clustering to the thrombospondin N-terminal region and the adjacent collagenous domain (Δ52, Δ53, Δ54, and Δ56 respectively), one heterozygous COL12A1 arginine-to-cysteine substitution of unclear significance (p.(Arg1863Cys)), and compound heterozygous pathogenic COL6A1 variants (c.[98–6G>A];[301C>T]) in one proband. Variant-specific intracellular accumulation of collagen XII chains, extracellular overmodification of the long isoform and near-absence of the short isoform of collagen XII, and extracellular decrease of decorin and tenascin-X were observed for the COL12A1 variants. In contrast, the COL6A1 variants abolished collagen VI and V deposition and increased tenascin-X levels.
Conclusion
Our data further support the significant clinical overlap between myopathic EDS and collagen VI–related myopathies, and emphasize the variant-specific consequences of collagen XII defects.
Similar content being viewed by others


INTRODUCTION
Collagen XII is a member of the fibril-associated collagen with interrupted triple helices (FACIT) family and primarily localizes to collagen I–rich tissues, such as ligaments, perichondrium, periosteum, dermis, and skeletal muscle.1 Collagen XII contains three α1(XII)-chains, encoded by the COL12A1 gene (OMIM ∗120320).2 Each chain consists of two helical collagenous domains (COL1 and COL2) flanked by two short carboxy-terminal noncollagenous domains (NC1 and NC2) and a substantial amino-terminal noncollagenous domain (NC3).3,4 NC3 consists of 18 fibronectin type III repeats, into which four von Willebrand factor A (vWA) domains are inserted, and a thrombospondin N-terminal (TSPN) domain.3,4 In the trimeric form, the NC3 domains fold into three arms, which extend from a stem formed by two short triple helical collagenous domains (Fig 1a). For the mammalian and avian α1(XII)-chain, a long and a short splice variant have been described, which differ by about 100 kDa in the length of their NC3 domain4 (Fig. 1b). These two isoforms can coexist in collagen XII trimers and are often coexpressed, although temporal and tissue-specific expression is observed.4 The long isoform is preferentially expressed during early embryonic development. At later developmental stages and in adult tissues the short isoform becomes the major form, while the long isoform continues to be expressed in a few dense connective tissues such as bone, tendon, and dermis.4,5,6 The isoforms also differ in their biochemical properties. The long isoform contains an additional heparin binding site in the seventh fibronectin type III domain and can occur as a proteoglycan, with covalently linked glycosaminoglycan chains.4

Collagen XII: domains and structure, variant location, myopathic Ehlers–Danlos syndrome (EDS) criteria and clinical characteristics. a The homotrimeric domain structure of collagen XII consists of two short triple helical domains (COL1 and COL2) separated by two short noncollagenous domains (NC1 and NC2) and large amino-terminal noncollagenous domains (NC3). NC3 is composed of a thrombospondin N-terminal (TSPN) domain, four von Willebrand factor A domains, and 18 fibronectin type III repeats. b Collagen XII variants described in this paper (upper five) and those reported up to date (lower six) are depicted on the collagen XII α1-chain. Collagen XII short splice variant is depicted in bright colors; the additional domains corresponding to the long splice variant are added as lighter symbols. Δ; exon skip in collagen XII. This figure is not drawn to scale. c Hypothetical model for the supramolecular assembly of interstitial fibrils in the dermis. Collagen XII is able to associate directly with fibril-forming collagens, probably via its two carboxy-terminal domains, COL1 and NC1.4,7 Its interaction partners regulate the distances between fibrils (collagen I and V) by direct (decorin, orange) or indirect (tenascin-X, blue) binding to collagen I-containing fibrils.30,31,32 The interaction of tenascin-X (blue) with the fibrils is mediated by decorin (orange).33 Collagen VI forms its own microfibrillar network. The tetramers align end-to-end in a unique microfibrillar pattern of thin and long aggregates in the dermis.34 An unknown protein (?, yellow) links collagen XII to collagen VI filaments via the small leucine-rich proteoglycans (SLRPs) decorin, and/or biglycan (orange), which preferentially form complexes in the vicinity of the amino-termini of collagen VI filaments.35 Collagen VI also interacts with the globular amino-terminal propeptide of collagen V, protruding on the surface of collagen I fibrils.27 In addition, fibronectin, which binds collagen I fibrils,36 is able to bridge the extracellular network to cell surfaces due to the interaction with specific integrin receptors.37 Other integrins can bind collagens. This figure is not drawn to scale. d Minimal criteria suggestive for myopathic EDS are major criterion 1 plus either one other major criteria and/or three minor criteria. e–g Clinical images of PIII:1 show a relatively mild phenotype. PIII:1 has a slender build (e), some atrophic scars (f), and micrognathia (g).
Various extracellular matrix (ECM) proteins interact with collagen XII via overlapping or distinct domains (Fig. 1c). Immunoelectron microscopy revealed that collagen XII associates with collagen I fibrils in tendon and skin, probably through its NC1 and COL1 domains.4,7 Collagen XII has also been shown to interact with the small leucine-rich proteoglycans (SLRPs) decorin and fibromodulin.8 Decorin interacts with the fibril-associating region of collagen XII through its glycosaminoglycan chain.8 The collagen XII NC3 domain strongly binds to tenascin-X, and both proteins colocalize in the dermis.9 More recently, collagen XII was shown to interact with cartilage oligomeric matrix protein (COMP) through its collagenous domains.10 Hence, collagen XII and its binding partners can form flexible bridges between collagen fibrils and other, noncollagenous matrix molecules, and as such are involved in regulation of the organization and mechanical properties of collagen fibrils in several tissues.
In 2014, two groups simultaneously reported homo- or heterozygous pathogenic variants in COL12A1 in individuals suffering from a mixed myopathic and Ehlers–Danlos syndrome (EDS)–like phenotype.6,11 Subsequently, two more families with a myopathy/EDS-like phenotype due to heterozygous COL12A1 variants were reported12,13 (Fig. 1b). These individuals displayed distal joint hypermobility in combination with proximal joint contractures, scoliosis or kyphosis, and abnormal scarring, and myopathic features including muscle hypotonia and weakness with delayed motor development.6,11,12,13,14 Based on the phenotype of the 13 hitherto reported patients, this condition was included in the recently revised EDS classification as myopathic EDS (mEDS)14 (Fig. 1d). In parallel with the human phenotype, Col12a1 knockout mice displayed variable muscle weakness with decreased grip strength, combined with signs of connective tissue abnormalities, such as bone fragility, short stature, and kyphoscoliosis.6,15 The muscle weakness present in this mouse model underscores the role of collagen XII in muscle tissue, thereby implying that structural changes in the myomatrix affect force transmission in the muscle tendon–bone unit.6
The clinical phenotype of mEDS strongly resembles that of mild myopathies that are caused by variants in COL6A1, COL6A2, or COL6A3, the genes encoding the three α-chains of the collagen VI heterotrimer. Defects in collagen VI result in a spectrum of myopathies, ranging from a relatively mild Bethlem myopathy (BM) (OMIM 158810),2 characterized by proximal muscle weakness and flexion contractures, to the more severe, progressive Ullrich congenital muscular dystrophy (UCMD) (OMIM 254090),2 characterized by congenital joint contractures and muscle hypotonia, but also connective tissue features such as kyphoscoliosis, hip dislocation and dysplasia, severe distal joint laxity, and abnormal wound healing and scarring.16,17 Despite the clinical overlap between collagen VI and collagen XII–related disorders, neither a direct nor an indirect molecular interaction between collagen VI and XII has been identified.
Here we report, among a cohort of 78 patients fulfilling the clinical criteria for mEDS, the identification of four novel heterozygous pathogenic COL12A1 variants in six patients from four families, one COL12A1 variant of unclear significance, as well as biallelic pathogenic variants in COL6A1 in an eighth patient. We show that these variants lead to variant-specific alterations in patient fibroblast cultures, both at the intracellular and extracellular level.
MATERIALS AND METHODS
Patient information and material
For this study, a cohort of 78 genetically unresolved patients who were referred to our center and fulfilled the clinical criteria for mEDS were selected (Fig. 1d, Table S1). The study was approved by the local Ethics Committee of the Ghent University Hospital, Belgium. PI:1–PVI:1 gave informed consent for the collection of a blood sample and skin biopsy; furthermore PIII:1 and her parents consented to the publication of clinical photographs. Blood samples were obtained from family members if possible. Genomic DNA (gDNA) was isolated from dermal fibroblast cultures or blood leukocytes using the PureGene extraction method (Gentra Systems, Minneapolis, MN, USA). Dermal fibroblast cultures from patients and age- and sex-matched controls were grown under standard conditions.
Variant analysis and sequencing
Coding exons and flanking intronic sequences of COL6A1 (NM_001848), COL6A2 (NM_001849), COL6A3 (NM_004369), and COL12A1 (NM_004370) were screened by next-generation panel sequencing (MiSeq, Illumina, San Diego, CA, USA). Variants were confirmed by bidirectional Sanger sequencing (ABI3730XL, Applied Biosystems, Foster City, CA, USA). Variant nomenclature is according to Human Genome Variation Society (HGVS) guidelines and pathogenicity was evaluated using the Alamut Visual software (version 2.3, Interactive Biosoftware). Reverse transcription polymerase chain reaction (RT-PCR) studies verified predicted splicing effects. Population frequency was assessed in the Genome Aggregation Database (gnomAD).18
Fibroblast analysis
Immunocytochemistry
For ECM studies, fibroblasts were seeded in duplicate in 8-well Nunc Lab-Tek chamber slides (Thermo Fisher Scientific, Waltham, MA, USA) and stimulated with 25 µg/ml ascorbate (Sigma-Aldrich, St. Louis, MO, USA) for 3 days. Cells were stained as described previously.6 For intracellular staining, cells were fixed 24 hours after seeding and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich). Images were taken of equally confluent areas with a Zeiss Axio Observer.Z1 microscope.
Western blotting
Confluent fibroblast cultures were incubated for 24 hours in serum-free medium and proteins were extracted in triplicate as described previously.19 Conditioned serum-free medium from fibroblast cultures was collected at day 14 as previously described.19 Protein samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) before blotting, either by wet blotting onto a polyvinylidene difluoride (PVDF) membrane for collagen XII and tenascin-X detection, or by dry blotting onto a nitrocellulose membrane for other proteins. Imperial protein staining (Perbio Science bvba, Erembodegem, Belgium) visualized the total protein amount. Imaging was performed on the Amersham Imager 680 (GE Healthcare Life Sciences) using ImageQuant software.
Both techniques were repeated thrice, using antibodies listed in Supplementary Data. Resulting images were processed with Fiji software.20
RT-qPCR
To compare the transcription levels of COL5A1, COL5A2, COL6A1, COL6A2, COL6A3, COL6A6, COL12A1, COL14A1, and DCN in fibroblasts from patients and controls, quantitative reverse transcription PCR (RT-qPCR) was performed as described previously.19 Total RNA was extracted in triplicate. Assays were performed in duplicate for each sample. qbase + 3.0 (Biogazelle, Zwijnaarde, Belgium) was used for data analysis, using reference genes HPRT1, YWHAZ, and RPL13A for normalization.
Transmission electron microscopy
A skin biopsy from the upper anterior arm of PII:1 was fixed and processed for ultrastructural analysis as described previously.21 Sections were viewed with a JEOL JEM 1400plus transmission electron microscope operating at 60 kV. The observations were compared with control samples of a six-month-old girl.
Statistical analysis
Data are expressed as mean ± standard error (SEM). P values less than 0.05 were considered significant. Statistical calculations, including multiple testing corrections, were performed using GraphPad Prism 5 (La Jolla, CA, USA).
RESULTS
Identification and clinical characterization of patients with novel COL12A1 and COL6A1 variants
Among 78 tested individuals, we identified five unique heterozygous COL12A1 variants, including four pathogenic splice-site variants and one missense variant of unclear significance (Table S2). One patient was compound heterozygous for two COL6A1 variants (Table S2).
In PI:1, a small genomic COL12A1 deletion c.8415+1_8415+10del resulted in in-frame skipping of exon 56 (Δ56). PII:1 harbors a COL12A1 c.8265+1G>A variant, leading to in-frame skipping of exon 54 (Δ54). In PIII:1, a COL12A1 c.8178+3A>C variant was found, causing in-frame skipping of exon 53 (Δ53). Segregation analysis revealed that the variant segregated in her father and her sister, who showed a much milder phenotype. In PIV:1, a small genomic COL12A1 deletion, c.8100+3_8100+6delGAGT, led to in-frame skipping of exon 52 (Δ52). These four splice-site variants are in close proximity to each other, either located in the COL2 domain (Δ56 and Δ54) or the adjacent TSPN domain (Δ53 and Δ52) (Fig. 1b). The pathogenicity of these variants is supported by their absence in population databases (gnomAD), the segregation of the variant in the third family, and the de novo occurrence in the other three patients (Table S2).
In PV:1, a heterozygous COL12A1 missense variant (c.5587C>T) was found, causing an arginine-to-cysteine substitution (p.(Arg1863Cys)), located in the 14th fibronectin type III domain in the large NC3 region of collagen XII (Fig. 1b). No family members were available for molecular analysis. The missense variant is in silico predicted to be pathogenic and is present in very low frequency in the gnomAD population database (Table S2).
In PVI:1, compound heterozygous variants in COL6A1 (c.[98–6G>A];c.[301C>T]) were identified (Fig. 4a). Segregation analysis revealed that the c.98–6G>A variant was inherited from the mildly affected mother (showing joint hypermobility, easy bruising, atrophic scarring), whereas the nonsense variant (c.301C>T, p.(Gln101∗)) was inherited from the healthy father. It is currently unclear whether the mild connective tissue signs present in the mother can be ascribed to her carrier status of a pathogenic variant. RT-PCR studies revealed that the intronic variant activates a cryptic acceptor splice site, adding four nucleotides to the beginning of the second exon and resulting in a frameshift (Fig. 4a). As such, both variants are predicted to produce truncated transcripts that are prone to nonsense-mediated messenger RNA (mRNA) decay (NMD).
Clinical findings are summarized in Table 1, details are provided in Supplementary Data, and compared with previously described patients in Table S3.
Consequences of COL12A1 variants on collagen XII protein levels and localization
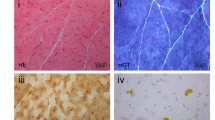
No differences in mRNA expression levels were noted on RT-qPCR for the long and short splice variants of COL12A1 between patient and control skin fibroblasts (data not shown). Nevertheless, western blotting (WB) revealed intracellular accumulation of α1(XII)-chains for patients PIII:1 (Δ53) and PIV:1 (Δ52), which was confirmed by immunocytochemistry (ICC) (Fig. 2). No significant intracellular accumulation was noted in PI:1 (Δ56), PII:1 (Δ54) and PV:1 (R1863C). ICC staining of extracellular collagen XII was normal in all analyzed probands (Fig. 2b). However, WB revealed abnormal migration patterns of secreted collagen XII in Δ56, Δ53, and Δ52 fibroblasts (Fig. 2a). The bands representing the long isoform of the monomeric α1(XII)-chain migrated slightly higher (lv1–330 kDa), as did the trimeric form composed of three long splice variant chains (lv3–990 kDa) and the dimer (lv2–660 kDa) for Δ56, Δ53, and Δ52, possibly due to overmodification. Furthermore, the bands representing the short splice variant (sv1–230 kDa), and the collagen XII trimeric combinations including one or more short splice variant(s) (three bands between 990 and 660 kDa representing trimers with one [lv2sv1], two [lv1sv2], or three [sv3] short isoforms respectively) were almost completely absent in Δ56, Δ53, and Δ52 (Fig. 2a). For Δ54 and R1863C, a normal migration pattern was detected.

Effects of COL12A1 variants on collagen XII protein levels and localization. a Western blotting of intracellular and extracellular collagen XII in patient fibroblasts compared with controls (C). Intracellular accumulation was observed in Δ53 and Δ52. Modified long splice variants (lv) α1(XII) are indicated with an arrow. In the medium, lack of the short splice variant (sv) (*) was noted in Δ56, Δ53, and Δ52. Loading control: β-tubulin for cell lysates, Imperial Blue gel staining for protein content in medium. b Immunocytochemistry of intracellular and extracellular collagen XII in patient fibroblasts compared with controls. For Δ53 and Δ52, intracellular accumulation of collagen XII was detected, similar to the results in (a). Secreted collagen XII appeared normal in all samples, but can show location-dependent variation in both patients and controls. Green channel: collagen XII signal. Blue channel: DAPI staining. Scale bar intracellular staining: 100 µm. Scale bar extracellular staining: 200 µm. Δ, exon skip in collagen XII; R1863C, p.(Arg1863Cys) variant.
Consequences of collagen XII defects on collagen XII interaction partners and on ECM organization
To determine if the identified COL12A1 variants affect protein levels of known interaction partners of collagen XII, we performed WB analysis of chondroitinase ABC (ChABC)–digested decorin. This revealed a strong reduction of decorin core protein levels in the medium of Δ56 and Δ53 and a milder reduction in Δ52 (Fig. 3a). WB analysis demonstrated significantly reduced levels of secreted tenascin-X in probands Δ54 and Δ53 and a trend toward reduction in Δ52 (Fig. 3a, b). ICC for tenascin-X and decorin failed due to lack of appropriate antibodies.

Investigation of ECM proteins and matrix structure. a Western blotting of secreted decorin and tenascin-X levels were compared between myopathic Ehlers–Danlos syndrome (mEDS) patients and matched control (C) samples. A reduction of extracellular decorin core protein was observed in Δ56, Δ53, and Δ52. Tenascin-X levels are clearly reduced in fibroblasts’ medium of Δ54, Δ53, and Δ52. b Due to the subtle differences in tenascin-X intensity via WB, relative quantities of tenascin-X levels in the extracellular matrix (ECM) of patient versus control were calculated from four different western blots with protein samples obtained at different time points. The tenascin-X levels were normalized against the loading control. A two-way analysis of variance (ANOVA) test was used to determine significance. c Cross-sectional transmission electron microscopy of the reticular dermis of Δ54 indicates a shift toward an increased collagen fibril diameter and shows increased interfibrillar spacing compared with a six-month-old control. Histogram of the collagen fibril diameter distribution and graph displaying the percentage of fibril surface and interfibrillar space measured on cross section (N = 590). d Elastin deposited in the reticular dermis of Δ54 shows a ruffled appearance on the edges. Data are expressed as mean ± SEM. A two-way ANOVA test was used to determine significance. **P < 0.01; ****P < 0.0001; Δ, exon skip in collagen XII; R1863C, p.(Arg1863Cys) variant.
WB and ICC studies with antibodies against collagen I, V, VI, and fibronectin, as well as RT-qPCR for COL5A1, COL5A2, COL6A1, COL6A2, COL6A3, COL6A6, COL14A1, and DCN did not show overt differences for patients compared with controls (data not shown). Furthermore, endoplasmic reticulum (ER) stress was not observed after WB analysis for ER stress markers BiP, eIF2A, phospho-eIF2A, and ATF6 (data not shown).
The ECM architecture was studied using transmission electron microscopy (TEM) of the reticular dermis in a skin biopsy sample from PII:1 (Δ54) and a sex-matched pediatric control biopsy (Fig. 3c). Morphometric analysis revealed mildly increased collagen fibril diameters in Δ54 (92.1 ± 0.3 nm) compared with the control biopsy (84.7 ± 0.3 nm). Interfibrillar spaces were filled with amorphous granulofilamentous deposits and were significantly larger (42 ± 1.44%) compared with control (32 ± 1.10%). Furthermore, the morphology of elastin showed an altered appearance with ruffled edges. Fibroblasts did not show clear dilation of the ER or autophagosome accumulation (data not shown).
Consequences of the COL6A1 defects on collagen VI protein levels and interaction partners
The predicted NMD of COL6A1 in PVI:1 (c.[98–6G>A];c.[301C>T]) was confirmed by RT-qPCR analysis, showing severely reduced COL6A1 expression in dermal fibroblasts (Fig. 4b). The mRNA levels of COL6A2 and COL6A3 transcripts were not affected (data not shown).

Molecular identification of COL6A1 defects. a The COL6A1 c.98–6G>A splice-site variant in the proband and the mildly affected mother of family 6 introduces a new splice acceptor site, resulting in the addition of four nucleotides to the transcript. The second collagen VI variant found in the proband and the unaffected father of family 6, COL6A1 c.301C>T (p.(Gln101*)), is a nonsense variant located in the amino-terminal vWA domain. b Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis of PVI:1 and two control individuals showed severely reduced COL6A1 expression in total RNA isolated from the affected individual’s skin fibroblast cultures. Data are expressed as mean ± SEM. A one-way analysis of variance (ANOVA) test was used to determine significance. c Biochemical electrophoretic analysis of collagen VI and tenascin-X from fibroblasts’ medium of patient and control (C). The absence of bands representing the α1/2(VI)- and α3(VI)-chain and increased secretion of tenascin-X in the patient was observed. d Extra- and intracellular immunocytochemical staining for collagen V and VI in patient and control fibroblasts revealed reduced staining at the extracellular level but no clear differences intracellularly. Extracellular images; red channel: collagen VI signal, green channel: collagen V signal. Intracellular images; green channel collagen VI and V signal respectively, blue channel: DAPI staining. ****P < 0.0001. Scale bar extracellular staining: 200 µm. Scale bar intracellular staining: 100 µm. mRNA messenger RNA.
WB for PVI:1 showed absence of the bands representing the secreted α1-, α2-, and α3(VI)-chains (Fig. 4c), while increased tenascin-X levels in the patient’s medium were observed (Fig. 4c).
ICC revealed absent staining for collagen V and VI at the extracellular level for PVI:1 in comparison with the control individual (Fig. 4d). No differences in intracellular staining for collagen V and VI were noted.
Collagen XII showed normal results on WB, ICC, and RT-qPCR, which was also the case for collagen I, collagen XIV, decorin, and fibronectin (data not shown).
DISCUSSION
Among a cohort of 78 patients fulfilling the clinical criteria for myopathic EDS, we identified seven patients from five families in whom we found novel heterozygous variants in COL12A1, and one patient with biallelic variants in COL6A1. This brings the total number of patients harboring pathogenic COL12A1 variants to 19, plus one patient with a COL12A1 variant of unclear significance. Salient clinical features among these patients include (1) (congenital) muscle hypotonia with variable delay in gross motor development, reduced muscle mass, muscle atrophy, and sometimes mild-to-moderate increase of serum creatine kinase (CK) levels; (2) joint hypermobility, which is either limited to distal joints, but which can also be generalized; (3) congenital and/or progressive joint contractures, including congenital talipes equinovarus, adducted thumbs and finger contractures, and sometimes contractures of larger joints; (4) mild involvement of the skin, with soft, smooth, pale, and/or translucent skin, mild delay in wound healing, and sometimes some atrophic or hypertrophic scarring; and (5) mild dysmorphic facial features, including light blue sclerae, micrognathia, and/or high arched palate. Other features, observed in few patients, include congenital torticollis, bilateral hip dislocation, scoliosis, kyphosis, pectus excavatum, and hyperkeratosis pilaris (Table S3).
To date, 11 COL12A1 variants have been identified in mEDS patients, including two glycine substitutions (p.(Gly2777Arg) and p.(Gly2786Asp)) and two splice-site variants causing in-frame skipping of exon 54 or 56, all located in the COL2 domain; three splice-site variants leading to the in-frame skip of either exon 52 or 53, all located in the TSPN domain; one p.(Ile2334Thr) substitution in a vWA domain; two arginine-to-cysteine substitutions located in the 14th (p.(Arg1863Cys)) and 15th (p.(Arg1965Cys)) FN type III domain of the NC3 domain, respectively; and one biallelic splice-site variant leading to an out-of-frame skip of exon 50, resulting in NMD and near-complete loss of collagen XII protein6 (Fig. 1b, Table S3). Notably, the splice-site variants cluster to the transition region between the NC3 domain and the fibril-associated region. This hinge region includes the TSPN and COL2 domain (Fig. 1a).
Based on the hitherto reported patients, some genotype–phenotype correlations emerge. Near-complete absence of collagen XII, due to biallelic pathogenic variants leading to NMD, is associated with the most severe phenotype, as demonstrated by the siblings reported by Zou et al., who had a very severe congenital phenotype, precluding independent ambulation.6 COL12A1 haploinsufficiency on the other hand seems to be associated with a very mild phenotype, illustrated by the parents of those siblings, who are obligate carriers and who only had some delay in early motor development. The in-frame exon skips and the amino acid substitutions lead to the production of qualitatively aberrant α1(XII)-chains, and are associated with variable, moderately severe phenotypes. Strikingly, the patients harboring an Arg-to-Cys substitution were diagnosed at an older age than the other mEDS patients11 and showed no joint hypermobility (Table S3). Both arginine residues are located in a FN type III domain, which normally does not contain cysteine residues (Figure S1). Introduction of a cysteine in this domain may result in abnormal disulfide bridge formations within or between α1(XII)-chains. In contrast to the p.(Arg1965Cys), p.(Arg1863Cys) did not show intracellular accumulation, nor reduced extracellular levels of collagen XII or other ECM proteins, and ER stress markers were normal (data not shown). It is possible that these arginine-to-cysteine substitutions are associated with milder phenotypes, and/or that they are disease-modifying, rather than disease-causing. Analysis of gnomAD data revealed the occurrence of substitutions in 91 of the 157 arginine residues throughout the α1(XII)-chain, occurring with varying frequencies (Figure S2). Twenty-three of those are substituted by a cysteine, but clinical information is not available for these individuals. Thorough phenotyping of other individuals with arginine collagen XII substitutions may give more insight into the pathogenicity of this variant type.
The patients’ skin fibroblasts with an exon skip in the TSPN domain (Δ52 and Δ53) showed intracellular accumulation of mutant collagen XII protein (Fig. 2), suggesting interference with secretion of the mutant protein. Intracellular collagen XII accumulation was previously also observed in fibroblasts of patients harboring another exon 52 skipping pathogenic variant,13 and in fibroblasts from the patients harboring the p.(Gly2777Arg), p.(Gly2786Asp), and p.(Arg1965Cys) substitutions.11,12
In all our patients, ICC showed an apparently normal collagen XII matrix, in contrast to the reduced or absent extracellular collagen XII in fibroblasts of all previously reported mEDS patients (Table S3). Possibly, differences in seeding density and protocol setup, in combination with a potentially decreased growing capacity of patient fibroblasts, influence collagen XII matrix formation, resulting in different observations. Nevertheless, WB revealed that the bands representing mono-, di-, or trimers of the long isoforms of α1(XII) migrated slightly higher in the medium of the patients with Δ52, Δ53, and Δ56, suggesting possible overmodification (Fig. 2a). Therefore we hypothesize that for these variants, incorporation of mutant chains may delay trimerization, allowing more time for intracellular modification of the long splice variant, and resulting in slower secretion and thus intracellular accumulation of the trimers containing the long isoform. In addition, we observed for these same variants decreased extracellular levels of collagen XII trimers containing one, two, or three short splice variants of α1(XII), compared with control. This suggests that secretion of trimers containing short isoform chains may be hampered by these specific exon skips. Alternatively, the aberrant splicing of these specific exons might interfere with the splicing mechanisms generating the short isoform, eventually leading to reduced levels of this isoform. We note that the in vivo situation, where the short isoform dominates, might differ from our observations since the long isoform predominates in cultured skin fibroblasts.5
To unravel possible consequences of collagen XII defects on the ECM architecture, (in)direct interaction partners were examined by ICC and WB (Fig. 1c). All patient dermal fibroblasts displayed normal ICC staining for collagen I, V, and fibronectin. Consistent with previous reports,6,11,12,13 collagen VI staining also showed no abnormalities for our patients, despite considerable clinical overlap with collagen VI–related myopathies (Table S3). Furthermore, ER stress was not observed in our patients. In contrast, decorin core protein levels were markedly reduced in patients with Δ52, Δ53, and Δ56, but not in Δ54 (Fig. 3a). The interaction of collagen XII with decorin is not fully understood, but decorin binds to an unspecified domain of the fibril-associated region through its glycosaminoglycan chain.8 Since decorin interacts with the N-terminal region of the thrombospondin-1 protein in skin fibroblasts,8,22 it could be hypothesized that decorin interacts with the TSPN domain of collagen XII, which may be affected in the patients with Δ52 and Δ53. Additionally, we observed a reduction of tenascin-X protein levels secreted from skin fibroblasts from patients with Δ52, Δ53, and Δ54 (Fig. 3b). Since tenascin-X binds to the NC3 domain of collagen XII,9 the exact consequences of these exon skips on the collagen XII–tenascin-X interaction are currently unclear. Possibly, these exon skips induce conformational changes in the hinge region and/or the NC3 domain, which may affect its interaction with tenascin-X.
TEM of the reticular dermis of PII:1 (Δ54) revealed a slight increase in collagen I fibril diameter and in interfibrillar space compared with control, consistent with a role for collagen XII in regulating collagen fibril diameter and packing.4 Of note, tendons of collagen XIV–deficient mice, another FACIT molecule, also showed a shift toward larger-diameter collagen fibrils.23 Furthermore, elastin in this patient seemed to have more ruffled edges and more microfibrils at the border. To our knowledge, a role for collagen XII in elastinogenesis has not yet been reported. Since PII:1 (Δ54) showed a strong reduction in extracellular tenascin-X levels, and since tenascin-X has been shown to negatively influence packing of dermal collagen fibrils24 and to localize to elastic fibers in vivo,25 it is conceivable that the tenascin-X decrease contributes to the observed ultrastructural changes in this patient.
In our cohort we also identified a young boy with compound heterozygous loss-of-function COL6A1 variants. As shown in Table 1, at the age of 2 years, his phenotype largely overlapped with that of the COL12A1 mEDS patients from our study, further underscoring the clinical overlap between collagen VI– and collagen XII–related conditions, at least early in life. However, clinical follow-up of this patient indicated progression of his phenotype toward a more classic UCMD. This is in contrast with the COL12A1 mEDS patients, who appear to improve over time.13 The latter observation will however need to be confirmed by long-term natural history studies in other mEDS patients. Both pathogenic COL6A1 variants in the COL6A1 proband are predicted to result in NMD, but residual mutant transcripts suggest that some escape NMD. Intracellular collagen VI staining did not show differences between patient’s fibroblasts and control, possibly due to staining of α2(VI)- and α3(VI)-chains. However, on WB α1(VI)-, α2(VI)-, and α3(VI)-chains were absent from the medium and collagen VI extracellular deposition was abolished, implying that α1(VI) is crucial in the formation of collagen VI heterotrimers. This was also observed in Col6a1−/− mice, which fail to generate triple helical collagen VI molecules due to the elimination of the α1(VI)-chain.26 Furthermore, no extracellular collagen V was observed, thereby emphasizing the important interaction between collagen V and VI.27 Contrary to the finding of reduced tenascin-X levels in some patients harboring COL12A1 pathogenic variants, secreted tenascin-X levels in this patient were elevated. This is consistent with transcriptome analyses on dermal fibroblasts of BM and UCMD patients and supports the suspected regulatory role of tenascin-X on collagen VI.28,29 ICC staining for collagen XII was normal, analogous to observations in earlier reported BM and UCMD patients.11,13
In conclusion, we report a novel α1(XII) arginine-to-cysteine substitution and four novel α1(XII) splice-site variants, leading to in-frame skipping of either exon 52, 53, 54, or 56, and expand the clinical and mutational spectrum of myopathic EDS. Most hitherto identified pathogenic COL12A1 variants cluster in the hinge region, underscoring its importance for proper collagen XII function and for interactions with other ECM molecules, notably decorin and tenascin-X. In addition, we report on a patient with compound heterozygous variants in COL6A1, whose early phenotype clinically resembles mEDS, further demonstrating the major clinical overlap between collagen XII– and collagen VI–associated conditions, at least in infancy. At the molecular level however, we note a clear distinction between both conditions, each affecting the ECM differently. Further functional studies in additional patients are necessary to understand variant-specific pathogenic mechanisms.
This study also underscores the power of targeted resequencing of gene panels, by means of next-generation sequencing techniques, to identify underlying genetic causes in patients who may present with mixed phenotypes. The majority (72/78) of our patients fulfilling the mEDS criteria however remain genetically unresolved, similar to the BM-like cohort reported by Hicks et al.11 This strongly suggests genetic heterogeneity within our cohort, although we cannot exclude deep intronic variants or copy-number variations in some. The limited number of patients with pathogenic COL12A1 variants described to date resulted in rather broad diagnostic criteria for mEDS.14 The identification of additional patients with COL12A1 variants may help to further demarcate mEDS from other genetic entities and fine-tune the clinical diagnostic criteria.
References
Gregory KE, Keene DR, Tufa SF, Lunstrum GP, Morris NP. Developmental distribution of collagen type XII in cartilage: association with articular cartilage and the growth plate. J Bone Miner Res. 2001;16:2005–2016.
McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604.
Gordon MK, Gerecke DR, Olsen BR. Type XII collagen: distinct extracellular matrix component discovered by cDNA cloning. Proc Natl Acad Sci USA. 1987;84:6040–6044.
Koch M, Bohrmann B, Matthison M, Hagios C, Trueb B, Chiquet M. Large and small splice variants of collagen XII: differential expression and ligand binding. J Cell Biol. 1995;130:1005–1014.
Oh SP, Griffith CM, Hay ED, Olsen BR. Tissue-specific expression of type XII collagen during mouse embryonic development. Dev Dyn. 1993;196:37–46.
Zou Y, Zwolanek D, Izu Y, et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet. 2014;23:2339–2352.
Keene DR, Lunstrum GP, Morris NP, Stoddard DW, Burgeson RE. Two type XII-like collagens localize to the surface of banded collagen fibrils. J Cell Biol. 1991;113:971–978.
Font B, Eichenberger D, Rosenberg LM, van der Rest M. Characterization of the interactions of type XII collagen with two small proteoglycans from fetal bovine tendon, decorin and fibromodulin. Matrix Biol. 1996;15:341–348.
Veit G, Hansen U, Keene DR, et al. Collagen XII interacts with avian tenascin-X through its NC3 domain. J Biol Chem. 2006;281:27461–27470.
Agarwal P, Zwolanek D, Keene DR, et al. Collagen XII and XIV, new partners of cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. J Biol Chem. 2012;287:22549–22559.
Hicks D, Farsani GT, Laval S, et al. Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy. Hum Mol Genet. 2014;23:2353–2363.
Punetha J, Kesari A, Hoffman EP, et al. Novel Col12A1 variant expands the clinical picture of congenital myopathies with extracellular matrix defects. Muscle Nerve. 2017;55:277–281.
Witting N, Krag T, Werlauff U, et al. Collagen XII myopathy with rectus femoris atrophy and collagen XII retention in fibroblasts. Muscle Nerve. 2018;57:1026–1030.
Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C. 2017;175:8–26.
Izu Y, Sun M, Zwolanek D, et al. Type XII collagen regulates osteoblast polarity and communication during bone formation. J Cell Biol. 2011;193:1115–1130.
Bonnemann CG. The collagen VI-related myopathies Ullrich congenital muscular dystrophy and Bethlem myopathy. Handb Clin Neurol. 2011;101:81–96.
Lamande SR, Bateman JF. Collagen VI disorders: insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018;71–72:348–367.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Syx D, Van Damme T, Symoens S, et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers–Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum Mutat. 2015;36:535–547.
Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682.
Van Damme T, Gardeitchik T, Mohamed M, et al. Mutations in ATP6V1E1 or ATP6V1A cause utosomal-recessive cutis laxa. Am J Hum Genet. 2017;100:216–227.
Merle B, Malaval L, Lawler J, Delmas P, Clezardin P. Decorin inhibits cell attachment to thrombospondin-1 by binding to a KKTR-dependent cell adhesive site present within the N-terminal domain of thrombospondin-1. J Cell Biochem. 1997;67:75–83.
Ansorge HL, Meng X, Zhang G, et al. Type XIV collagen regulates fibrillogenesis: premature collagen fibril growth and tissue dysfunction in null mice. J Biol Chem. 2009;284:8427–8438.
Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501.
Egging DF, van Vlijmen I, Starcher B, et al. Dermal connective tissue development in mice: an essential role for tenascin-X. Cell Tissue Res. 2006;323:465–474.
Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140.
Symoens S, Renard M, Bonod-Bidaud C, et al. Identification of binding partners interacting with the alpha1-N-propeptide of type V collagen. Biochem J. 2011;433:371–381.
Paco S, Casserras T, Rodriguez MA, et al. Transcriptome analysis of Ullrich congenital muscular dystrophy fibroblasts reveals a disease extracellular matrix signature and key molecular regulators. PLoS ONE. 2015;10:e0145107.
Butterfield RJ, Dunn DM, Hu Y, Johnson K, Bonnemann CG, Weiss RB. Transcriptome profiling identifies regulators of pathogenesis in collagen VI related muscular dystrophy. PLoS ONE. 2017;12:e0189664.
Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729–743.
Hedbom E, Heinegard D. Interaction of a 59-kDa connective tissue matrix protein with collagen I and collagen II. J Biol Chem. 1989;264:6898–6905.
Brown DC, Vogel KG. Characteristics of the in vitro interaction of a small proteoglycan (PG II) of bovine tendon with type I collagen. Matrix. 1989;9:468–478.
Elefteriou F, Exposito JY, Garrone R, Lethias C. Binding of tenascin-X to decorin. FEBS Lett. 2001;495:44–47.
Engel J, Furthmayr H, Odermatt E, et al. Structure and macromolecular organization of type VI collagen. Ann N Y Acad Sci. 1985;460:25–37.
Wiberg C, Hedbom E, Khairullina A, et al. Biglycan and decorin bind close to the N-terminal region of the collagen VI triple helix. J Biol Chem. 2001;276:18947–18952.
Ruoslahti E, Hayman EG, Kuusela P, Shively JE, Engvall E. Isolation of a tryptic fragment containing the collagen-binding site of plasma fibronectin. J Biol Chem. 1979;254:6054–6059.
Pytela R, Pierschbacher MD, Ruoslahti E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell. 1985;40:191–198.
Acknowledgements
We thank the patients and their families for their contribution to the study. Special thanks to Prof. Manuel Koch (University of Cologne) for providing us the anti-collagen XII (KR75) and anti-tenascin-X antibodies. DS, FM and OV are postdoctoral fellows of the Research Foundation Flanders (FWO), Belgium. This work was supported by a Methusalem Grant (BOFMET2015000401) from the Ghent University to Prof. Anne De Paepe.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Delbaere, S., Dhooge, T., Syx, D. et al. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet Med 22, 112–123 (2020). https://doi.org/10.1038/s41436-019-0599-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0599-6
Keywords
This article is cited by
-
The Non-pregnant and Pregnant Human Cervix: a Systematic Proteomic Analysis
Reproductive Sciences (2022)
-
A multi-ethnic genome-wide association study implicates collagen matrix integrity and cell differentiation pathways in keratoconus
Communications Biology (2021)
-
The Ehlers–Danlos syndromes
Nature Reviews Disease Primers (2020)
