
Abstract
Purpose
Maternal diabetes is a known teratogen that can cause a wide spectrum of birth defects, collectively referred to as diabetic embryopathy (DE). However, the pathogenic mechanisms underlying DE remain uncertain and there are no definitive tests to establish the diagnosis. Here, we explore the potential of DNA methylation as a diagnostic biomarker for DE and to inform disease pathogenesis.
Methods
Bisulfite sequencing was used to identify gene regions with differential methylation between DE neonates and healthy infants born with or without prenatal exposure to maternal diabetes, and to investigate the role of allele-specific methylation at implicated sites.
Results
We identified a methylation signature consisting of 237 differentially methylated loci that distinguished infants with DE from control infants. These loci were found proximal to genes associated with Mendelian syndromes that overlap the DE phenotype (e.g., CACNA1C, TRIO, ANKRD11) or genes known to influence embryonic development (e.g., BRAX1, RASA3). Further, we identified allele-specific methylation (ASM) at 11 of these loci, within which 61.5% of ASM single-nucleotide variants are known expression quantitative trait loci (eQTLs).
Conclusions
Our study suggests a role for aberrant DNA methylation and cis-sequence variation in the pathogenesis of DE and highlights the diagnostic potential of DNA methylation for teratogenic birth defects.
Similar content being viewed by others


INTRODUCTION
Teratogenesis is the disruption of normal fetal development consequent to an environmental exposure. Pregestational diabetes is a well-known teratogen, and infants of diabetic mothers (IDMs) are two to fourfold more likely to develop birth defects compared with the general population.1,2 Likewise, gestational diabetes (GDM) is also associated with increased risks of birth defects.3,4 As a result, ~4–9% of IDMs are born with diabetic embryopathy (DE)1,2—a phenotypic spectrum of birth defects that overlaps recognizable genetic and genomic syndromes.4 This creates a diagnostic challenge, as the presence of multiple birth defects often triggers an expensive diagnostic odyssey aimed at providing information regarding recurrence risk, future medical management, and recommendations for disease screening. Currently, however, there is no definitive clinical diagnostic test for diabetic exposure, DE, or any other teratogenic exposure.
DNA methylation, mostly found at cytosines in cytosine–guanine dinucleotides (CpGs), is a malleable regulatory epigenetic feature that can reflect prenatal exposures to environmental stimuli, such as maternal nutrition.5 Maternal diabetes in humans might thus disrupt normal epigenetic patterns, resulting in aberrant gene function during embryogenesis, and ultimately lead to malformations in the fetus. Previous studies of DNA methylation in IDMs have focused on epigenetic changes that could influence cardiometabolic risk in later life;6,7,8,9,10,11,12,13 however, such studies provide only one view of methylation following diabetic pregnancies and none assessed DNA methylation in the context of DE.
Given this gap in knowledge and the emergence of studies using methylation signatures to support the diagnosis of congenital syndromes,14,15,16 we used genome-wide capture bisulfite sequencing, which interrogates >5 million CpG sites, to assess DNA methylation in buccal samples from neonates with congenital malformations and a history of prenatal diabetes exposure. Buccal cells were chosen over blood, a commonly used, but more heterogeneous surrogate tissue, partly to facilitate sampling and analyses, but also because buccal methylation is more consistently correlated with methylation in other (nonblood) cells;17 thus our results potentially also reflect methylation patterns in other tissues. Our starting premise was that teratogenic effects incurred during early embryogenesis will mirror Mendelian disorders by being substantial in magnitude and affecting all germ layers. Given the fidelity of mitotic DNA methylation inheritance, we hypothesized that traces of these effects would still be evident in differentiated tissues around the time of birth. In so doing, we aimed to gain insight into the underlying pathogenesis and evaluate the diagnostic potential of DNA methylation for DE.
MATERIALS AND METHODS
Participants
We recruited 22 infants through Texas Children’s Hospital in Houston, Texas, representing the IDM phenotypic extremes (Table S1): 9 had a diagnosis of DE based on the presence of maternal diabetes during pregnancy and congenital malformations consistent with that diagnosis, 6 IDMs were completely healthy at birth (exposed, healthy; EH), and the remaining 7 were healthy infants born to healthy mothers (unexposed, healthy; UH). Table S2 lists malformations, genetic tests, as well as detailed infant and maternal demographics. At enrollment, mothers of infants with DE had significantly higher body mass indices (BMIs) compared with healthy mothers (p = 0.017, Tukey honestly significant difference [HSD]; Table S1, Fig. S1), making maternal obesity an unavoidable confounder of diabetes exposure. The remaining demographics were not significantly different between groups (Table S1).
The study was approved by the Institutional Review Board at Baylor College of Medicine (H32571). Written informed consent was provided by the parents of all subjects.
Sample collection
Buccal epithelial cells were collected within ten days of birth using the Oragene Discover (OGR-250) DNA collection kit (DNA Genotek Inc., Ottawa, Ontario, Canada), modified to obtain a cumulative buccal sample by doing five passes per cheek with each of five swabs. DNA extraction, sequencing, methylation quantification,18 and genetic variant analysis19,20 are described in the Supplementary Methods. Samples were sequenced at one timepoint, but in two batches: the first (discovery) batch comprised three UH, one EH, and four DE infants; the second (replication) batch consisted of three UH, four EH, and three DE samples, excluding outliers (see below). Samples from both batches were combined in our joint analysis.
Postsequencing processing and quality control
Analyses were performed using only CpG sites with ≥10× coverage in every sample (median: 26×). Potential batch effects were evaluated using one-way analysis of variance (ANOVA) on the distribution of medians and quantro software (1000 data permutations; version 1.4.0) (ref. 21). There was no strong evidence of batch effects (p = 0.544, F(1,11) = 0.392, one-way ANOVA; quantro permutation p = 0.597), nor were there other statistically significant biases in the distribution of cases, or infant or maternal demographics (Table S3). Based on a tissue prediction algorithm,22 all samples were confirmed to be primarily buccal in origin (Table S4, Supplementary Methods).
Given the breadth of clinical phenotypes in the cohort, a principal component analysis (PCA) and hierarchical clustering of samples was performed. Four outlier samples—one UH, one EH, and two DE infants (Fig. S2)—were subsequently removed. This conservative approach assumes that most CpG sites are not differentially methylated between cases and controls. After outlier removal, 2,800,516 CpG sites were shared between UH and DE samples.
Differential methylation analysis
Coverage normalization and single site differential methylation analysis using a logistic regression model without covariates was performed using methylKit (version 0.9.5) (ref. 23). To reduce false positives in a manner agnostic to the data distribution, all sites with nominal p values < 0.001 were permuted 1000 times with replacement by randomly switching case/control labels. Adjacent high-confidence differentially methylated loci—sites at which ≤10 permutations produced an equally small or smaller p value than the original analysis—were then clustered into bins. All high-confidence sites within a bin were separated from neighboring sites by no more than 1 kb and shared the same direction of effect. Bins that did not contain at least one high-confidence differentially methylated site with an absolute difference in percent methylation ≥10 were excluded, as were those that did not contain at least one CpG site found to be differentially methylated (p < 0.05, ≤10/1000 better or equal permutations) using a secondary software, methylSig (version 0.4.1) (ref. 24), which assesses differential methylation using a β-binomial approach. Post hoc power analysis showed that our study had 93.7% power at ɑ = 0.05 (Supplementary Methods).
Classification
To test classification on the joint batch data set we applied fourfold cross-validation with ten repetitions (Supplementary Methods) using the default settings of WEKA’s “Logistic” classifier (version 3.6.13) (ref. 25). To assess the utility of methylation patterns for assigning cases and controls when blinded to sample labels, we repeated the differential methylation analysis using only the discovery batch, with two exceptions to account for the smaller sample size: a bin size of ≥7 high-confidence sites (Fig. S3) and decreased ɑ of 0.01 in methylSig analysis. Classification was performed on high-confidence sites found in the largest bins using WEKA’s logistic regression model with default settings, including a ridge parameter of 10−8, one iteration, and no scaling (coefficients, intercept, and odds ratios are listed in Table S5). As a secondary approach, we adapted the correlation-based classification method used by Choufani et al.14 by labeling test instances as cases or controls depending on whether their Pearson correlation was stronger with the discovery cases or controls, respectively.
RESULTS
Differential methylation distinguishes clinical outcomes among diabetes-exposed and unexposed infants
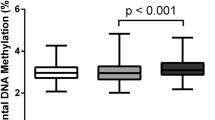
After removing four outliers (2 DE, 1 EH, and 1 UH; Fig. S2), we evaluated buccal DNA methylation at 2,760,543 CpG sites shared between all infant groups. Global mean percent methylation was lower in DE infants compared with UH controls (42.6% vs. 43.2%; p = 0.045, Tukey HSD; Fig. 1a). This reduction in global methylation occurred predominantly at CpG loci that, compared with UH control samples, typically had high levels of methylation (>70%; p = 0.011, Tukey HSD; Fig. 1b).

Infants with diabetic embryopathy (DE) show global DNA hypomethylation compared to unexposed, healthy controls (UH). Boxplots show the distribution of mean sample percent methylation across the number of sites in each category. (a) 2,760,543 sites shared among DE, UH, and diabetes-exposed, yet healthy infants (EH); (b) 1,362,136 (49.3%) sites with mean methylation <30% in UH infants; 276,215 (10.0%) sites with mean methylation 30–70% in UH infants; 1,122,192 (40.7%) sites with mean methylation >70% in UH infants. Lines in density plot represent sample group means across the distribution. *Tukey honestly significant difference (HSD) p < 0.05.
Global epigenetic differences among DE infants suggested that the effect of diabetes exposure on the methylome was substantial; therefore, we considered whether sites with the largest effects might be useful biomarkers of the maternal diabetes exposure. A logistic regression model revealed 13,239 high-confidence sites (0.5% of total sites) that surpassed our stringent, permutation-based cut-off and thus had the strongest evidence of differential methylation (p < 0.001) between UH and DE. We then leveraged the anticipated correlations between neighboring CpG loci to further eliminate spurious associations by clustering high-confidence sites into nonoverlapping bins. Differentially methylated sites fell into 237 bins (≥3 high-confidence differentially methylated sites per candidate bin, each containing at least one significant (p < 0.05) CpG site using our secondary method). Consistent with our global analysis, the majority (87.8%) of these bins were hypomethylated (Table 1, Table S6). The mean absolute difference in percent methylation across all 1010 high-confidence CpG sites in these bins was 16.6 (SE 0.3).
Using PCA on all 1010 binned high-confidence CpG sites, we found that UH samples clustered separately from infants with DE (Fig. 2a). Moreover, when we repeated the same analysis including EH infants, they showed a clustering that was separate from, and intermediate to, DE and UH (Fig. 2b). The resulting heat map of differentially methylated sites further bolstered this observation—EH individuals demonstrated an intermediate pattern to UH and DE, with most sites having a comparable magnitude of differential methylation (Fig. 2c). Fourfold cross-validation correctly categorized samples with 100% accuracy, suggesting that the differential methylation patterns were distinct enough to be exploited for classification.

Distinct DNA methylation profile in diabetic embryopathy (DE) cases compared with controls. Each analysis was based on all sites found in candidate bins with ≥10× coverage and non zero variance across samples. (a) Principal component analysis (PCA) based on 1010 high-confidence differentially methylated CpG sites shared across all DE and unexposed, healthy control (UH) infants. (b) PCA based on 1001 high-confidence differentially methylated CpG sites shared across all DE, UH, and diabetes-exposed, yet healthy (EH) infants. Difference in numbers between a and b is the result of coverage adequacy in EH samples. (c) Heat map of scaled and centered percent methylation values corresponding to b, with each row representing one high-confidence differentially methylated site, and each column a sampled individual. (d) Heat maps of scaled and centered z-scores in DE and mean z-scores of 10,000 sample permutations in public data set and in other clinical conditions (e), including autism spectrum disorder (ASD), Down syndrome (DS), and fetal alcohol spectrum disorder (FASD). Each row represents one high-confidence differentially methylated site also found in public array data and each column a participant. (f) Boxplots of sample mean percent methylation at high-confidence CpG sites from the differentially methylated cluster coinciding with CACNA1C. (g) Heat map and hierarchical clustering of scaled and centered percent methylation for CpGs at CACNA1C locus.
Next, we compared DE methylation patterns at our candidate loci with those of 29 publicly available age- and tissue-matched, healthy controls whose DNA methylation had been analyzed with the Illumina 450 K array.26 Because our targeted-bisulfite sequencing approach probed substantially more loci than the 450 K array, we compared data sets based on z-score distributions at the 72 differentially methylated CpG sites that overlapped both platforms (Supplementary Methods). Despite the limited CpGs, methylation in DE infants was distinct from that in publicly obtained healthy controls (Fig. 2d), but was indistinct from controls if we conducted the same analysis using an equal number of loci without evidence of differential methylation (Fig. S4A). Subsequently, we repeated the z-score comparison with publicly available, buccal cell 450 K data from 84 individuals with developmental and congenital disorders, including autism spectrum disorder (ASD),27 Down syndrome (DS),28 and fetal alcohol spectrum disorder (FASD).16 Using 87 of our differentially methylated CpG loci captured across data sets, we found that DE samples were also distinct from samples with other disorders (Fig. 2e) and this was not evident using an equal number of nonsignificant CpG loci (Fig. S4B). Our results implied that the magnitude of deviation from control DNA methylation levels observed in DE is not only unusual for our cohort, but also atypical compared with a larger set of independently sampled healthy neonates and other syndromic conditions. Lastly, CpG sites in the peripheral blood DNA methylation signatures of other syndromes14,15 could not distinguish DE from healthy controls on PCA, implying that the DE malformations have a methylation signature distinct from those of other syndromes (Figs. S5–7).
Differentially methylated sites as diagnostic classifiers of infants with DE
We then further explored whether highly differentially methylated sites could be used to blindly classify clinical status. Of the 3,092,753 shared CpG sites analyzed in our discovery batch of four DE and three UH samples, 13,731 high-confidence sites (0.4%) showed evidence of differential methylation (p < 0.001) and surpassed our stringent permutation-based cut-off. These sites were further filtered to 15 high-confidence bins containing at least seven differentially methylated loci. High-confidence bins had an average absolute difference in percent methylation of 28.6 (SE 2.0) between DE and UH infants and again the majority (73.3%) were hypomethylated in DE infants.
Using 156 CpG sites in our high-confidence bins with adequate coverage in our test batch, a logistic regression model could classify the remaining six UH and DE test samples with 100% accuracy (6/6). Wary of overfitting, we applied a second, correlation-based classifier, which distinguished UH from DE samples in 5/6 (83.3%) instances (Fig. 3a). To investigate how EH infants would be grouped using a classifier trained to distinguish DE from UH individuals, we applied both methods to the 155 CpG sites shared by all infants. The logistic regression model classified 2/5 (40%) of EH samples as “DE,” thereby performing no better than random; however, the correlation-based model labeled 100% of EH samples as “DE,” suggesting that this method might better distinguish between diabetes-exposed and unexposed individuals.

Batch classification results. Predictions in green were classified correctly in the comparisons between diabetic embryopathy (DE) and unexposed, healthy control (UH) (a) and between DE and diabetes-exposed, yet healthy (EH) (b) samples. Of the CpG loci used to distinguish between sample groups in each comparison, nine were present in both (c).
Not all pregnancies exposed to maternal diabetes result in birth defects; therefore, we evaluated the potential to discern malformation from exposure by comparing the two groups with prenatal exposure to maternal diabetes in the same classifier analysis. Because most EH samples were sequenced in the second batch, we carefully matched them to the demographic characteristics of DE individuals in the two data sets (test of batch effects: p = 0.769, F(1,10) = 0.091, one-way ANOVA; quantro permutation p = 0.673; Table S3). Differential methylation analysis and permutation testing revealed 19,139 (0.6% of 3,197,718 loci) high-confidence differentially methylated sites, of which 197 fell into 21 high-confidence bins. In contrast to the previous comparison, fewer than half of these bins were hypomethylated in DE relative to EH individuals (10/21, 47.6%). Both classification methods correctly identified 80% (4/5) of samples, although the misidentified individuals differed between methods (Fig. 3b). Similar classification of IDM samples alongside buccal array data from ASD, DS, and FASD showed comparable promise (Supplementary Table S11).
Only nine9 differentially methylated sites distinguished infants with DE from both UH and EH neonates (Fig. 3c), all of which were contained in the same bin and were hypermethylated in DE infants. This region is less than 4.5 kb upstream of CSF3 (chr17: 38,167,214—38,167,428)—a granulocyte colony stimulating factor that, in rodents, has been shown to protect against left ventricular remodeling and cardiac myocyte apoptosis after myocardial infarction.29
IDM-related differential methylation overlaps developmental genes
To further characterize the changes in DNA methylation and their potential relationship to the clinical presentation of diabetic embryopathy, we interrogated the gene content of the differentially methylated sites from the initial joint analysis. Differentially methylated sites were particularly enriched for intronic (28.7% versus 24.5%; p < 0.001), and intergenic (37.0% versus 25.9%; p < 3.52 × 10−15) annotations (Fig. S8), which have varying functional consequences on gene regulation.30
Of the loci coinciding with genes associated with genetic disorders, the bin overlapping an intronic region of CACNA1C showed a pronounced distinction between DE infants and healthy controls based on both mean sample methylation (Fig. 2f) and hierarchical clustering (Fig. 2g). Heterozygous loss-of-function variants in CACNA1C, which encodes a voltage-dependent calcium channel subunit, cause Timothy syndrome (MIM 601005). The distinct loss of methylation at this locus in DE infants in conjunction with the gene’s cardiac involvement and the prominence of cardiac phenotypes in IDM (Table S2) cautiously suggest a potential role for CACNA1C in DE.
In addition to CACNA1C, we observed differential methylation at other Mendelian disease genes, including ANKRD11, the causal gene in KBG syndrome (MIM 148050; Fig. S9A), which includes spinal and digit malformations as well as occasional heart defects; B3GNT1, associated with Walker–Warburg syndrome (MIM 615287); BRF1, which results in the characteristic central nervous system (CNS) and skeletal abnormalities of cerebellofaciodental syndrome (MIM 616202); TRIO, associated with autosomal dominant mental retardation (MIM 617061), microcephaly, and digit malformations;31 and ZBTB20, which causes the large stature noted as part of Primrose syndrome (MIM 259050). Differential methylation was also observed at other genes that result in DE-like features when knocked out in mouse models, including BARX1 (embryonic lethal with cleft palate;32 Fig. S9B) and RASA3 (abnormal embryogenesis including abnormal vascular endothelial cell development33). In addition, we found an expansion of the DE-characteristic region upstream of CSF3, which was second in the number of binned CpG sites only to another hypermethylated region encompassing two microRNAs of unknown function, MIR3648 and MIR3687 (Table 1, Fig. S9C).
To get a more agnostic sense of the genes implicated in our study, we performed pathway enrichment and gene ontology analyses using all genes within 1 kb of binned regions (n = 176; Supplementary Methods).34 Two of six enriched pathways (nominal p ≤ 0.01) were related to cardiac and neuron function (phase 2: plateau phase and DCC mediated attractive signaling; Table S7)—both major systems affected in DE. Among the 24 enriched (nominal p ≤ 0.01) gene ontology terms, ten related to basic cellular and organismal development including actin function, ion binding, cell development, and nervous system function and development (Table S8).
Allele-specific methylation suggests a role for sequence variation in diabetic embryopathy
Finally, we decided to leverage the genetic information from our bisulfite sequencing data to evaluate the influence of sequence variation on the observed methylation differences (Supplementary Methods). To avoid ambiguity over whether single-nucleotide variants (SNVs) were true or artifacts of the bisulfite conversion process, we focused on individuals homozygous for reference and alternate alleles. We identified 609 pairs of plus-stranded CpG cytosines and common SNVs that were homozygous in at least two samples and within 100 bp of the methylation locus (Fig. 4a). Of these pairs, 62 (10.1%) showed evidence of allele-specific methylation (ASM), defined in our analysis as a mean difference in percent methylation between reference and alternate alleles outside of the sample distribution (<5th centile; >95th centile). Eleven (4.6%) of the 237 bins differentially methylated in DE infants contained at least 1 of 24 ASM events among our candidate loci (Table S9). These events overlapped six intergenic regions and five RefSeq genes: CDH12, ECHDC3, NTM, RNF157, and TRIO. Eight (61.5%) of these 13 SNVs are known expression quantitative trait loci (eQTLs) that are specific to certain tissues, such as the cerebellum, or are found across many different cell types (Table S10).

Allele-specific methylation (ASM) at differentially methylated CpG loci. (a) Mean % methylation of homozygous reference vs. homozygous alternate allele samples. Each genotype category contains at least two individuals. Colored points are found within clustered regions of differential methylation. Yellow dots show one single-nucleotide variant (SNV) associated with ASM at two proximal CpG sites found in an intergenic region (b). Pink dots show one SNV associated with ASM at four proximal CpG sites found in TRIO (c).
Among the pairs with the most pronounced difference in percent methylation were two CpG sites associated with the variant rs142180914 in an intergenic region, which showed increasing DNA methylation with increasing numbers of reference alleles (Fig. 4b). Remarkably, the only samples homozygous for the alternate allele were from infants with DE. We also observed four CpG loci that were strongly correlated with the intronic variant rs389729 in TRIO, but showed a decrease in methylation for each additional reference allele (Fig. 4c). In this case, all individuals homozygous for the alternate allele were diabetes-unexposed controls. Although neither rs142180914 nor rs389729 are currently known to affect gene expression, the high proportion of eQTLs among our SNV-CpG pairs with evidence of ASM at differentially methylated loci suggests that common genetic variants could contribute to an individual’s predisposition to develop DE upon prenatal exposure to maternal diabetes.
DISCUSSION
The known teratogenic effects of maternal diabetes, the absence of confirmative diagnostic tests for DE, and the rising prevalence of diabetes reinforce the need to improve diagnostics for DE. Our work demonstrates that neonates with DE have altered DNA methylation profiles compared with healthy newborns. The identified DNA methylation changes, implicating developmental genes and known Mendelian disorders, could guide future studies of the yet unresolved disease pathogenesis. Furthermore, a selection of informative CpG loci show promise as biomarkers for a diagnostic test of DE, the diagnosis of which currently relies on the exclusion of phenotypically similar conditions.
Other studies have already shown the utility of DNA methylation to improve diagnoses of congenital disorders, such as Sotos syndrome (MIM 117550) and Weaver syndrome (MIM 277590),14 as well as CHARGE (MIM 214800) and Kabuki (MIM 147920) syndromes.15 Similar to our investigation, a recent study of fetal alcohol spectrum disorder—caused by a teratogenic exposure to ethanol—also identified altered DNA methylation patterns in buccal epithelial cells.16 Comparable with the aforementioned Mendelian disorders, our results suggest that the teratogenic effect causing DE has a large influence on offspring DNA methylation and could allow for accurate distinction between UH and DE infants using modest sample sizes. Using PCA, we could also distinguish diabetes-exposed, but healthy infants from diabetes-exposed infants with congenital malformations. Thus, DNA methylation biomarkers may also inform the severity of the teratogenic effect. A screening test based on DNA methylation patterns could be used to efficiently evaluate the likelihood of diabetes exposure as the cause of birth defects among IDMs—an option that does not presently exist.
We were careful in our analysis to focus on large differences in methylation that passed stringent statistical thresholds of significance; however, we did not include covariates such as maternal smoking, which has been associated with altered DNA methylation in offspring.35 The methylation patterns of the only (UH) infant in our differential methylation analysis whose mother smoked during pregnancy appeared unremarkable, suggesting that maternal smoking did not substantively influence our results. DE was marginally associated with maternal obesity, which has been shown to influence offspring DNA methylation36 and is an independent, albeit less pronounced, risk factor for birth defects. Given that obese women are more likely to develop T2DM and GDM, it remains to be seen whether the teratogenic mechanisms of maternal obesity are distinct from or synergistic with those of maternal diabetes.
The results presented here still require a broader assessment of DNA methylation for the diagnosis of DE to generate robust estimates of sensitivity and specificity. Recruitment of additional samples, including infants with birth defects but no diabetes exposure, IDMs with less severe complications, and older children/adolescents further removed from their prenatal exposure, would facilitate clinical implementation. As long as the DNA methylation fingerprint is consistently correlated with the phenotype, accessible tissues, such as buccal epithelial cells, are sufficient (and even preferred) for participant sampling; however, formal evaluation of methylation patterns across cell types is needed to fully understand whether the changes in DNA methylation seen in buccal epithelial cells are consistent and causally implicated in DE.
Maternal diabetes has also been associated with increased reactive oxygen species (ROS) levels in the fetus.37,38 DNA damage incurred by ROS is known to prompt cell cycle arrest and induce apoptosis, yet, even a cell that has escaped apoptosis can be left with substantial damage. Given that double-strand break repair can produce heritable changes in DNA methylation at the break locus,39 the repair mechanisms invoked by ROS damage could result in similar DNA methylation disruptions, which might significantly alter the expression of important developmental genes. Our gene and pathway analyses converged upon a number of genes affecting embryonic development, including many underlying known genetic syndromes; this could explain the strong phenotypic overlap between many of these syndromes and DE.
Nonrandom alterations in DNA methylation patterns could, at least in part, be explained by genetic influences. We identified SNVs that correlated with DNA methylation at proximal CpG sites. The majority of SNVs found at differentially methylated loci are known eQTLs in a variety of tissues. Differential methylation occurring at CpG sites under the control of cis-genetic variation could be the result of differences in the chance distribution of ASM alleles between cases and controls; alternatively, this might reflect genetic variation that, upon exposure to maternal diabetes, potentiates changes in DNA methylation in an allele-specific manner and predisposes an individual to DE.
In light of the emerging role of DNA methylation and accompanying cis-regulatory variation across a spectrum of environmental exposures,40 our framework of epigenetic aberrations and cis-genetics may be applicable to other teratogenic exposures, paving the way for improvements in diagnostics, and ultimately therapeutics, for this class of disorder.
References
Sheffield JS, Butler-Koster EL, Casey BM, McIntire DD, Leveno KJ. Maternal diabetes mellitus and infant malformations. Obstet Gynecol. 2002;100:925–930.
Sharpe PB, Chan A, Haan EA, Hiller JE. Maternal diabetes and congenital anomalies in South Australia 1986-2000: a population-based cohort study. Birth Defects Res A Clin Mol Teratol. 2005;73:605–611.
Garcia-Patterson A, et al. In human gestational diabetes mellitus congenital malformations are related to pre-pregnancy body mass index and to severity of diabetes. Diabetologia. 2004;47:509–514.
Schaefer-Graf UM, et al. Patterns of congenital anomalies and relationship to initial maternal fasting glucose levels in pregnancies complicated by type 2 and gestational diabetes. Am J Obstet Gynecol. 2000;182:313–320.
Dominguez-Salas P, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746.
Allard C, et al. Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics. 2015;10:342351.
Ruchat SM, et al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics. 2013;8:935–943.
Petropoulos S, et al. Gestational diabetes alters offspring DNA methylation profiles in human and rat: identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology. 2015;156:2222–2238.
Finer S, et al. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24:3021–3029.
Haertle L, et al. Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin Epigenetics. 2017;9:28.
West NA, Kechris K, Dabelea D. Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism. 2013;1:1–9.
Gautier JF, et al. Kidney dysfunction in adult offspring exposed in utero to type 1 diabetes is associated with alterations in genome-wide DNA methylation. PLoS ONE. 2015;10:e0134654.
del Rosario MC, et al. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metabolism. 2014;63:654–660.
Choufani S, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207.
Butcher DT, et al. CHARGE and Kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet. 2017;100:773–788.
Lussier AA, et al. DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin Epigenetics. 2018;10:5.
Lowe R, et al. Buccals are likely to be a more informative surrogate tissue than blood for epigenome-wide association studies. Epigenetics. 2013;8:445–454.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572.
Liu Y, Siegmund KD, Laird PW, Berman BP. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13:R61.
McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Hicks SC, Irizarry RA. quantro: a data-driven approach to guide the choice of an appropriate normalization method. Genome Biol. 2015;16:117.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Akalin A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87.
Park Y, Figueroa ME, Rozek LS, Sartor MA. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics. 2014;30:2414–2422.
Hall M, et al. The WEKA data mining software: an update. ACM SIGKDD Explorations Newsletter. 2009;11:10–18.
Martino D, et al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14:R42.
Berko ER, et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014;10:e1004402.
Jones MJ, et al. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med Genomics. 2013;6:58.
Harada M, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11:305–311.
Neri F, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77.
Pengelly RJ, et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet, 2016;53:735–742.
Miletich I, et al. Developmental stalling and organ-autonomous regulation of morphogenesis. Proc Natl Acad Sci USA. 2011;108:19270–19275.
Iwashita S, et al. Versatile roles of R-Ras GAP in neurite formation of PC12 cells and embryonic vascular development. J Biol Chem. 2007;282:3413–3417.
Kamburov A, et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–717.
Breton CV, et al. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–467.
Sharp GC, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2015;44:1288–1304.
Wei D, Loeken MR. Increased DNA methyltransferase 3b (Dnmt3b)-mediated CpG island methylation stimulated by oxidative stress inhibits expression of a gene required for neural tube and neural crest development in diabetic pregnancy. Diabetes. 2014;63:35123522.
Zabihi S, Loeken MR. Understanding diabetic teratogenesis: where are we now and where are we going? Birth Defects Res A Clin Mol Teratol. 2010;88:779–790.
Russo G, et al. DNA damage and repair modify DNA methylation and chromatin domain of the targeted locus: mechanism of allele methylation polymorphism. Sci Rep. 2016;6:33222.
Teh AL, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014;24:1064–1074.
Acknowledgements
The authors thank Nancy Hall for invaluable technical help with sample management. This research was supported by a Baylor College of Medicine Presidential Award (J.W.B.) and USDA Agricultural Research Service (ARS) cooperative agreement (58–3092–5–001). N.A.H. was supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award (grant number 2013096). K.V.S. was supported by award number T32GM008307 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
J.W.B. is a full-time employee of Illumina Inc., but all work relevant to the current study was performed whilst an employee of Baylor College of Medicine. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Schulze, K.V., Bhatt, A., Azamian, M.S. et al. Aberrant DNA methylation as a diagnostic biomarker of diabetic embryopathy. Genet Med 21, 2453–2461 (2019). https://doi.org/10.1038/s41436-019-0516-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0516-z
Keywords
This article is cited by
-
Edematous severe acute malnutrition is characterized by hypomethylation of DNA
Nature Communications (2019)
