
Abstract
Purpose
Heritable factors play an important etiologic role in connective tissue disorders (CTD) with vascular involvement, and a genetic diagnosis is getting increasingly important for gene-tailored, personalized patient management.
Methods
We analyzed 32 disease-associated genes by using targeted next-generation sequencing and exome sequencing in a clinically relevant cohort of 199 individuals. We classified and refined sequence variants according to their likelihood for pathogenicity.
Results
We identified 1 pathogenic variant (PV; in FBN1 or SMAD3) in 15 patients (7.5%) and ≥1 likely pathogenic variant (LPV; in COL3A1, FBN1, FBN2, LOX, MYH11, SMAD3, TGFBR1, or TGFBR2) in 19 individuals (9.6%), together resulting in 17.1% diagnostic yield. Thirteen PV/LPV were novel. Of PV/LPV-negative patients 47 (23.6%) showed ≥1 variant of uncertain significance (VUS). Twenty-five patients had concomitant variants. In-depth evaluation of reported/calculated variant classes resulted in reclassification of 19.8% of variants.
Conclusion
Variant classification and refinement are essential for shaping mutational spectra of disease genes, thereby improving clinical sensitivity. Obligate stringent multigene analysis is a powerful tool for identifying genetic causes of clinically related CTDs. Nonetheless, the relatively high rate of PV/LPV/VUS-negative patients underscores the existence of yet unknown disease loci and/or oligogenic/polygenic inheritance.
Similar content being viewed by others


INTRODUCTION
Hereditary connective tissue disorders (HCTDs) comprise a large group of diseases with various manifestations affecting the skin, eyes, internal organs (e.g., the lung and intestine), as well as the neurological, craniofacial, musculoskeletal, and cardiovascular systems.1 Although conditions are individually rare, together they represent a considerable health burden.2 Several HCTDs are complicated by severe, partially life-threatening vascular complications such as aortic dilatations, aneurysms, dissections, or arterial tortuosity.3 HCTDs with vascular involvement include both syndromic forms such as Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS), and vascular Ehlers–Danlos syndrome (EDS) and nonsyndromic presentations of thoracic aortic aneurysm/dissection (TAAD). Together, these conditions are referred to as hereditary aortopathies.4 Three main pathomechanisms have emerged for hereditary aortopathies: (1) abnormal extracellular matrix assembly and maintenance/homeostasis, (2) increased TGF-β signaling, and (3) disruption of the vascular smooth muscle cell contractile apparatus.5,6 Accordingly, most known genes implicated in hereditary aortopathies encode structural connective tissue proteins such as collagens and fibrillins, enzymes involved in the biosynthesis or processing of structural connective tissue proteins (e.g., PLOD1), TGF-β signaling proteins (e.g., TGFBR1, SMAD3), or regulators and components of the cytoskeleton (e.g., MYH11).3 Connective tissue is found in different organs, thus a genetic alteration in a single HCTD-related disease gene can manifest in several body systems. This pleiotropy results in clinically overlapping multisystemic findings, which hampers clinical diagnosis of a specific disorder in the majority of patients.1 However, patient management including proper surveillance, timely surgical interventions, and pharmacological therapy depends on accurate diagnosis; this is already reflected in guidelines on the diagnosis and treatment of aortic diseases belonging to HCTDs.7,8 Genetic testing provides information that enables or supports an accurate diagnosis, which can help to define the medical course of action and, furthermore, to identify other at-risk family members.3 Due to pronounced and still growing heterogeneity, conventional consecutive disease/candidate gene screenings for the highly heterogeneous group of TAAD-related connective tissue disorders are both time- and cost-intensive; moreover, the high rate of patients negative for a causative sequence variant suggests that several risk genes are still unknown.3 To shorten analysis time, minimize costs, and increase detection rates, next-generation sequencing (NGS)-based genetic testing strategies, either targeted NGS (tNGS) or exome sequencing (ES), for aortopathies and related disorders of connective tissue have been applied recently and, overall, described as a tool that overcomes problems intrinsic to consecutive disease/candidate gene screening.9,10,11,12 Detection rates of causative sequence variants in 7 to 21 disease genes tested were between 3.9% and 35.3%, thus, in more than 60% of the patients, no causative variant was identified.9,10,11,12,13,14,15,16 To date, more than 20 disease genes for aortopathies, e.g., LDS, MFS, and nonsyndromic TAAD, have been identified;3 moreover, disease genes for several HCTDs with facultative cardiovascular manifestations, such as congenital contractural arachnodactyly (CCA), cutis laxa type IB, and arterial tortuosity syndrome, have been established.1 With a special focus on aortopathies and vascular manifestations, we selected 32 known disease genes and genetically tested a mixed cohort of 199 patients with the clinical diagnosis of hereditary aortopathy and/or related disorder of connective tissue by using NGS technology. Here we report on our experiences with this NGS-based molecular test as an obligate part of the diagnostic clarification of this group of disorders over a two-year period.
MATERIALS AND METHODS
Study cohort
We recruited patients in various medical centers in Germany (described in Supplementary Materials and Methods). Inclusion criteria were as follows: (1) Clinical features of the probands were suggestive of MFS, LDS, EDS, CCA, syndromic/nonsyndromic TAAD, or an unspecified heritable disorder of connective tissue with or without vascular involvement, i.e., HCTD. (2) Clinical data and samples for all individuals were obtained with informed consent of the patients’ parents/legal guardians or the patients themselves according to the Declaration of Helsinki and the national legal regulations (e.g., the German Genetic Diagnosis Act [GenDG]). In total, we obtained blood samples from 199 consecutive, unrelated patients with a mean age of 42 (±15.6 SD) years from January 2016 to December 2017. Characteristics of the study group including initial diagnoses, associated structural abnormalities, family history, etc. are described in the Supplementary Materials and Methods and summarized in Table S1.
Genetic testing
Selection criteria for the 32 genes and strength of gene–disease associations are outlined in Supplementary Materials and Methods and Table S2, respectively. Detailed information on targeted next-generation sequencing, exome sequencing, Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), transcript analysis, and statistical analysis are given in the Supplementary Materials and Methods and in Tables S2 and S3.
Variant prioritization, pathogenicity assignment and refinement
Synonymous, missense, and nonsense variants; coding indels; and intronic alterations at exon–intron boundaries ranging from −40 to +40 were included in our analysis. HCTDs with vascular involvement are relatively late-onset diseases and unlikely to have a huge effect on fitness and hence allele frequencies; therefore, variants with minor allele frequencies (MAF) ≥ than the respective disease prevalence were excluded (Table S4). Variants passing these filters were assigned to pathogenicity classes according the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) standards and guidelines: pathogenic variant (PV), likely pathogenic variant (LPV), variant of uncertain significance (VUS), and likely benign variant (LBV).17,18 Population and disease-specific databases, in silico predictive algorithms, as well as additional lines of evidence used for defining accurate classification criteria for variants are described in the Supplementary Materials and Methods. Variants with discordant classifications (d.c.) among this study and database on the relationships between human variations and phenotypes (ClinVar)19 or the Human Gene Mutation Database (HGMD)20 entries as well as variants with classifications having been modified after passing the ACMG/AMP classification system were categorized as refined variants (Table S5). Reasons for refinement are specified.
RESULTS
Identified variants by classification
We analyzed 199 individuals with hereditary aortopathy or related connective tissue disorder. The majority of patients had vascular complications such as TAAD, carotid artery aneurysm and dissection (CAAD), abdominal aortic aneurysm (AAA), or intracranial aneurysms (IA). One hundred fifty-two patients were tested by tNGS and 47 were screened by ES. In total we found 111 variants with MAF < the respective disease prevalence (Table S4) in 24 of 32 genes analyzed (Table S2). No variants were detected in COL1A1, COL4A5, EFEMP2, MAT2A, MFAP5, PLOD1, SLC2A10, and SMAD4. According to ACMG/AMP guidelines for variant classification,17,18 we defined 15 pathogenic variants (PV), 20 likely pathogenic variants (LPV), and 72 variants of uncertain significance (VUS) (in total 107 reportable variants) (Table 1, S6, and S7). Details on criteria for classifying variants are given in Table 1 and S5–S8; the types of variants identified are summarized in the Supplementary Results.
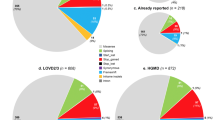
Four sequence alterations were classified as likely benign (LBV) and excluded from further data evaluation (Table S8). Eleven of the 15 PV were already annotated as (likely) disease-causing variants or (likely) pathogenic in HGMD or ClinVar, the other 4 PV were novel (Fig. 1). Of the variants classified as LPV, 11 were already reported, and 9 were novel (Fig. 1). The majority of PV/LPV were detected in FBN1 (23 variants) followed by SMAD3 (5 variants), TGFBR1 (2 variants) and FBN2, COL3A1, LOX, MYH11, and TGFBR1 (1 variant each) (Fig. 1). Of the 72 VUS, 25 were novel and 47 were already annotated in HGMD, ClinVar, or gnomAD; most VUS were identified in DCHS1 (12), COL3A1 (7), NOTCH1 (7), FBN2 (6), and FBN1 (5) (Fig. 1).

Summary of reportable variants identified by gene. The distribution of reportable variants including pathogenic variants (PVs), likely pathogenic variants (LPVs), and variants of uncertain significance (VUS) identified in genes associated with hereditary aortopathies and related disorders of connective tissue across the Hamburg cohort of 199 individuals is shown. Numbers of PV, LPV, and VUS per gene are given. Among the 107 reportable variants, 15 were pathogenic (4 novel), 20 were likely pathogenic (9 of these novel), and 72 were VUS (25 previously unreported).
For the novel variant FBN1 c.1589–14A>G (subject 14), we experimentally analyzed the impact on precursor messenger RNA (pre-mRNA) splicing because activation of a cryptic splice acceptor site in intron 13 was predicted (Table 1). We identified FBN1 transcripts with a 13-nucleotide 5’-expansion of exon 14 specifically in complementary DNA (cDNA) from patient 14, which results in a frameshift and a premature stop codon (p.Ile531Phefs*6) (Figure S1). Accordingly, FBN1 c.1589–14A>G was classified as LP (Table 1). In total, impact on splicing was predicted for 30 variants, which then were considered for variant classification according Richards et al. (Table 1 and S5–S8).17 For the classification of seven variants (i.e., by assigning BS4/PP1 attributes),17 we determined familial segregation (Figure S2). ACTA2 c.420G>A did not cosegregate with the disease in the family of subject 95 (Figure S2A). There was also no cosegregation of the COL3A1 variant c.805G>C with the disease in the family of subject 151, however, FBN2 c.1610A>G did cosegregate with the disease (Figure S2B). Affected but not healthy family members of subject 124 share the heterozygous FBN1 c.1211C>G variant (Figure S2C). Results for the remaining variants were not informative and, therefore, not included in variant classification (Figures S2D and S2E).
We screened PV/LPV-negative patients with a clinical diagnosis of MFS, LDS 1/2, and vascular EDS for copy-number variations in FBN1, TGFBR1, TGFBR2, and COL3A1, respectively, by MLPA; no alteration was detected.
LOX variants
We uncovered LOX variants in two patients by retrospective analysis of available ES data: the 23-year-old patient 188 with negative family history, aortic aneurysm, and dissection but negative for syndromic features, has the LPV LOX c.125G>A (p.Trp42*); in addition this patient carries a VUS in FBN2 (Table 1 and S6). Subject 13, 48 years old and with negative family history, initially was diagnosed with LDS based on an aortic aneurysm, translucent skin, easy bruising, hernia, and recurrent injuries of ankle ligaments; he underwent an ascending aorta replacement with reconstruction of the native aortic valve and reimplantation of the coronary arteries. This individual carries LOX c.995A>G (p.Tyr332Cys), which was classified as a VUS (PM1+PM2+BP4). Taken together, these two cases demonstrate the advantage of ES over tNGS because of the possibility to reanalyze existing ES data of mutation-negative patients and evaluate newly reported disease-associated genes; furthermore, ES data can be used to identify genes not yet associated with TAAD/HCTD.
Variant refinement
By using ACMG guidelines we obtained 12 and 14 discordant variant classifications for in total 21 variants compared with HGMD and ClinVar, respectively (Table S5). Ten HGMD classifications were downgraded (7 from DM to LPV, 1 from DM to VUS and 2 from DM[?] to VUS), and 2 were upgraded from DM(?) to PV (Table S5). Four ClinVar classifications were downgraded (3 from P to LPV and 1 from VUS to LBV), and 10 were upgraded from LB and VUS to VUS and LPV/PV, respectively (Table S5).
We also refined two classifications determined by ACMG guidelines. First, FBN2 c.1610A>G identified in subject 151 was reclassified from VUS (PM1+PP3) to LPV (PM1+PP1moderate+PP3+PP4, Table 1) because it cosegregates with the disease in four affected family members (Figure S2B) and the differential diagnosis for this patient was CCA. Second, LOX c.125G>A (p.Trp42*) was initially classified as VUS (PM2). However, this variant has been identified in two affected family members with familial TAAD; moreover another nonsense variant (p.Gly202*) has been described in a patient with familial TAAD.21 Thus, although not yet established, evidence for loss-of-function (LOF) pathogenicity is available,22 and we refined the LOX c.125G>A to be LP (PVS1strong+PM2).
Taken together, variant refinement resulted in up- or downgrading of 22 variants; thus, 19.8% of all detected variants (22 refined variants/111 variants in total) were reclassified.
Identified variants by patients
In 118 individuals (59.3%) we did not identify reportable variants with MAFs less than the respective disease prevalence; 81/199 patients (40.7%) were positive for reportable variants. Several patients carry more than one PV, LPV, and/or VUS, which explains the difference between the number of variants and the number of patients with reportable variants. Table S9 summarizes the distribution of reportable variants (PV, LPV, and VUS) by patient and the 25 individuals with more than one PV, LPV, or VUS are listed in Table S10. This data raises the question of the relevance of additional variants in the pathogenesis of HCTD.
Notably, four patients (14, 126, 142, and 162) had a variant in both FBN1 and NOTCH1 (Table S10), however, the phenotype of the four patients was neither atypical, extraordinarily severe, nor did the patients show a bicuspid aortic valve (clinical details are described in the Supplementary Results). Similarly, patients 97, 138, 151, 161, and 188 carry one VUS in addition to a PV/LPV (Table S10); the clinical manifestations in the five patients were not significantly different from the typical clinical spectrum of the respective disorder (clinical details are described in Supplementary Results). Summarized, our data suggest that concomitant reportable variants had a minor or even no contribution to the clinical outcome of the affected individuals.
Diagnostic yields
We identified at least one PV or LPV in 34 patients of our cohort resulting in an overall diagnostic yield of 17.1% (34/199 patients) (Table 2).
The highest diagnostic yield, 29.8% (17/57), was obtained in the subgroup of patients with the clinical diagnosis MFS (Table 2). In 16.9% (12/71) and 6.6% (4/61) of patients diagnosed with TAAD and HCTD, respectively, a PV/LPV was found (Table 2). The diagnostic yield was not significantly increased in patients with a positive family history compared with those with a negative family history (18.2% [16/88] vs. 13.9% [10/72]; p = 0.523) (Table 2). Next, we compared the initial clinical diagnoses between patients with and without PV/LPV in at least one of the analyzed 32 genes. The frequency of the initial diagnosis MFS was significantly higher in the PV/LPV-positive (17/34) compared with the PV/LPV-negative (40/165) cohort (50.0% vs. 24.2%; p = 0.0059) (Table 2). In contrast, the initial diagnoses HCTD and TAAD were not increased in the PV/LPV-positive subcohort (Table 2). Data and calculations for diagnostic yields considering VUS in addition to PV/LPV are presented in the Supplementary Results and Table S11. Taken together, there is a higher a priori probability to identify a causative variant in a patient with the clinical diagnosis MFS than TAAD and HCDT.
DISCUSSION
Diagnostic yield
Although the knowledge of the genetic causes for hereditary aortopathies and connective tissue disorders is constantly growing, many affected individuals still lack a genetic diagnosis. NGS-based multigene testing is currently regarded as the most powerful technology for genetic testing in clinical settings. In the cohort tested here by using NGS, 17.1% were positive for variants classified as P/LP. For individuals with the initial diagnosis MFS, vascular EDS, LDS, or CCA we identified at least one PV/LPV in 18/67 patients, thus the diagnostic yield for syndromic forms of HCTDs with vascular involvement is 26.9%. Several studies on NGS applications for the molecular diagnosis of TAAD have been reported.9,10,11,12,13,14,15,16 Taking into account only PV and LPV, the diagnostic yields were documented as 3.9% (21 genes in 102 patients analyzed),12 4.9% (15 genes in 1025 patients analyzed),13 8.1% (21 genes in 810 patients),16 10.3% (10 genes in 175 patients analyzed),11 13% (7 genes in 264 patients analyzed),15 18.6% (11 genes in 70 patients analyzed),14 27% (14 genes in 55 patients analyzed),10 and 35.3% (10 genes in 51 patients analyzed).9 In our subcohort of 71 patients with the initial diagnosis of TAAD, 12 individuals had a PV or LPV resulting in a diagnostic yield of 16.9%. Given the relatively permeable inclusion criteria resulting in a phenotypically diverse study cohort, the total diagnostic yield of 17.1% is decent. It was suggested that differences of the diagnostic yields may be caused among others by differing inclusion criteria.9,10 Indeed, high or increased diagnostic yields were observed when patients with a positive family history, with early-onset aortic disease, or with a syndromic form of TAAD were included primarily.9,10,13 In our study, however, the diagnostic yield was not increased in patients with a positive family history. Furthermore, in three previous studies the cohorts mainly included individuals with an unambiguous TAAD phenotype; however diagnostic yields were not consistent.12,13,14 Factors influencing the diagnostic yield may be different variant classification rules. First, we used phenotype prevalences as MAF cutoffs, whereas rather lenient/arbitrary maximum MAFs of 0.1% or 0.5% have been considered in other studies.9,10,12,13 Second, we interpreted variants according to the recommendations of the ACMG/AMP,17,18 which was also done in previous studies.11,13,14 A less stringent variant classification may result in a higher number of P/LP variants and, thereby, in an increased diagnostic yield.9,10 Third, for several variants the pathogenicity predictions strongly differ depending on the tools used.23 We used three ensemble scoring methods, REVEL, M-CAP, and ClinPred, each of them integrating scores from several individual tools (such as SIFT, PolyPhen, etc.); ensemble scoring methods constantly outperform individual tools.23,24,25,26 In contrast, various combinations of pathogenicity prediction tools, mostly individual tools, have been applied in previous studies.9,10,11,12,13,14,15,16 Taken together, standardization of patient inclusion criteria, variant classification, and pathogenicity prediction may improve study comparability and comprehensiveness.
The relatively low diagnostic yields (<< to <40%) observed in all related studies with an average of 15.4% indicate that in many patients the disease is caused by factors that are at present unknown.9,10,11,12,13,14,15,16 These can be genetic factors in coding or noncoding regions, which may exert their pathogenic potential in a mono-, oligo-, or polygenic manner. The latter inheritance pattern can include (rare, uncommon, and common) variants with relatively high MAF (e.g., >0.5%); those have not been considered in this study.
It has been assumed that inclusion of additional genes in the panels will increase diagnostic yields.9,11 In our study the number of analyzed genes was extended by at least 11 compared with previous studies,9,10,11,12,13,14,15,16 however, diagnostic yield was not consistently increased; in detail, we identified only one LPV (in addition to 18 VUS) in previously not-analyzed genes. This suggests that, on the assumption of a monogenic trait, pathogenic variants in yet undescribed or unknown disease genes may explain only a small fraction of HCTD/TAAD each.3,27,28 Clearly, nongenetic, i.e., environmental and lifestyle factors, may also account for a considerable number of cases with TAAD, and these certainly limit the diagnostic yield of genetic testing.
Clinical sensitivity and variant classification
Variant classification is a crucial step in NGS-based diagnostics that determines clinical sensitivity, i.e., the true positive rate. Re-evaluation of known variants will help to verify or falsify variant classifications, thereby uncovering more and more true genetic risk factors and improving clinical sensitivity. Refinement of variants in our study resulted in up- or downgrading of 22 variants based on ACMG/AMP classification criteria such as independent reports, segregation data, pathogenicity predictions, novel functional data, concomitant PV/LPV, transcript analysis, or the patient’s phenotype. For the same reasons variants have been reclassified in other studies.11,13,14 In this context, the consequences of variants on pre-mRNA splicing should be determined experimentally or at least by using prediction tools, as it is also recommended by the ACMG guidelines.17 In addition to increasing the diagnostic yield, in our study the assessment of consequences on splicing particularly helped to discriminate between VUS and LBV. Taken together, there is and there will be no perfect a priori variant classification system; rather valid classifications strongly depend on continual a posteriori refinement of variants, and this in turn requires ongoing data sharing within the scientific community and incorporation of refined classification results into databases.
Genotype and phenotype
Similar to our results, pathogenic and likely pathogenic variants in ACTA2, COL1A1, COL3A1, COL5A2, FBN1, NOTCH1, MHY11, MYLK, SLC2A10, TGFBR1, and TGFBR2 were the most abundant in TAAD cohorts.10,12,13,16 These results may reflect the well-described involvement of the reported genes in the pathogenesis of aortopathies and related disorders of connective tissue.29 On the other hand, these genes (and their products) are better characterized than most of the other genes tested, which likely contributed to the predominance of PV/LPV in these genes. Thus, variants in other genes may be underestimated and underclassified due to limited available evidence for pathogenicity. For example, the LOX c.995A>G (p.Tyr332Cys) variant identified in patient 13 was classified as VUS based on ACMG guidelines.17 It affects the LOX catalytic domain that covers all pathogenic missense variants reported to date.21,30 This domain contains ten fully conserved cysteine residues, which all are involved in disulfide bonds and render extracellular LOX an unusually stable enzyme.31 Thus, the change of tyrosine for a further cysteine has high potential to disturb the structure of the LOX catalytic domain, thereby, suggesting pathogenic consequences for the p.Tyr332Cys variant. Similarly, the pathogenicity of FBN2 variants identified in our cohort may also be underestimated (details are outlined in the Supplementary Discussion).
In 17/57 patients with the clinical diagnosis of MFS, the identification of a PV/LPV in FBN1 confirmed the clinical diagnosis. We did not detect a PV/LPV in FBN1 in 40 (70.2%) of 57 individuals with the initial clinical diagnosis of MFS. In keeping with this, in a recent study 40% of patients with a PV/LPV in FBN1 had a known or suspected clinical diagnosis of MFS.13 These data strongly differ from detection rates for pathogenic FBN1 variants of >90% in patients fulfilling a clinical diagnosis of MFS based on the Ghent nosology.32,33 In clinical practice MFS or MFS-like often is suspected for clinically ambiguous cases who have heritable aortopathy (i.e., TAAD) but do not obviously have MFS. Thus, the diagnostic pipeline should a priori include genetic analyses, thereby integrating the benefits of molecular genetics testing for the growing heterogeneity and pleiotropy of HCTD with vascular involvement. Given the highly variable expressivity and the marked phenotypic overlap observed in connective tissue disorders with vascular involvement, we and others conclude that a more lenient clinical evaluation of patients followed by an obligate stringent multigene analysis may help to avoid misdiagnoses.11
Notably, 25 individuals were found to carry more than one PV, LPV, or VUS. Concomitant variants in different genes in patients with TAAD have been reported previously, and the difficulty of variant classification and prioritization in these cases was highlighted.14,34 Severe clinical manifestations have been described in three patients with concomitant variants in different genes;14 however, it remained unclear whether multiple variants in different genes predict disease severity. Here, we describe four patients with reportable variants each in FBN1 and NOTCH1 but no atypical or severe clinical features. This suggests minor or no relevance of the identified NOTCH1 variants in the pathogenesis of the disease and/or a bias for overclassification of NOTCH1 variants, which may result from pronounced intolerance to variation/loss of function (missense Z Score = 3.95, pLI Score = 1.00) of NOTCH1.
Summary and conclusion
We applied NGS-based gene test approaches in a clinically relevant cohort of sporadic and familial individuals with hereditary aortopathies and related disorders and identified PV/LPV in 17.1% of patients. The highest diagnostic yield (29.8%) was obtained for patients with the clinical diagnosis of MFS. The lower detection rates for PV/LPV in patients with TAAD (16.9%) and HCTD (6.6%), and in parallel the higher incidence of VUS in these subcohorts suggest the existence of yet unknown disease loci and/or oligogenic/polygenic inheritance. In total 23.6% of PV/LPV-negative patients had VUS in genes associated with connective tissue disorders; these variants have to be re-evaluated in the future. Ultimately, gene-specific functional assays, RNA splicing studies, and further genetic analyses such as genome sequencing and/or genetic burden tests are needed to provide further evidence for the pathogenicity of variants, thereby increasing both the diagnostic yield and the clinical sensitivity. For the patients’ benefits, refining variant classifications will strongly support counseling of affected individuals as well as shaping therapeutic strategies.
References
Murphy-Ryan M, Psychogios A, Lindor NM. Hereditary disorders of connective tissue: a guide to the emerging differential diagnosis. Genet Med. 2010;12:344–354.
Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet. 2009;10:173–183.
Verstraeten A, Luyckx I, Loeys B. Aetiology and management of hereditary aortopathy. Nat Rev Cardiol. 2017;14:197–208.
Goyal A, Keramati AR, Czarny MJ, Resar JR, Mani A. The genetics of aortopathies in clinical cardiology. Clin Med Insights Cardiol. 2017;11:1179546817709787.
Andelfinger G, Loeys B, Dietz H. A decade of discovery in the genetic understanding of thoracic aortic disease. Can J Cardiol. 2016;32:13–25.
Morisaki T, Morisaki H. Genetics of hereditary large vessel diseases. J Hum Genet. 2016;61:21–26.
Erbel R, Aboyans V, Boileau C, et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2873–2926.
Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–369.
Poninska JK, Bilinska ZT, Franaszczyk M, et al. Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: diagnostic yield, novel mutations and genotype phenotype correlations. J Transl Med. 2016;14:115.
Proost D, Vandeweyer G, Meester JA, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat. 2015;36:808–814.
Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, et al. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet A. 2015;167A:1747–1757.
Ziganshin BA, Bailey AE, Coons C, et al. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015;100:1604–1611.
Weerakkody R, Ross D, Parry DA. et al. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet Med. 2018;20:1414–1422.
Fang M, Yu C, Chen S, et al. Identification of novel clinically relevant variants in 70 Southern Chinese patients with thoracic aortic aneurysm and dissection by next-generation sequencing. Sci Rep. 2017;7:10035.
Campens L, Callewaert B, Muino Mosquera L, et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities—results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis. 2015;10:9.
Overwater E, Marsili L, Baars MJH, et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum Mutat. 2018;39:1173–1192.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Biesecker LG, Harrison SM. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018;20:1687–1688.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067.
Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581.
Guo DC, Regalado ES, Gong L, et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118:928–934.
Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;99:247.
Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017;18:225.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885.
Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586.
Alirezaie N, Kernohan KD, Hartley T, Majewski J, Hocking TD. ClinPred: prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am J Hum Genet. 2018;103:474–483.
Vanakker O, Callewaert B, Malfait F, Coucke P. The genetics of soft connective tissue disorders. Annu Rev Genomics Hum Genet. 2015;16:229–255.
Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. 2016;133:2516–2528.
Brownstein AJ, Ziganshin BA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA. Genes associated with thoracic aortic aneurysm and dissection: an update and clinical implications. Aorta (Stamford). 2017;5:11–20.
Lee VS, Halabi CM, Hoffman EP, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A. 2016;113:8759–8764.
Chen X, Greenaway FT. Identification of the disulfide bonds of lysyl oxidase. J Neural Transm (Vienna). 2011;118:1111–1114.
Baetens M, Van Laer L, De Leeneer K, et al. Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat. 2011;32:1053–1062.
Loeys B, De Backer J, Van Acker P, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24:140–146.
Sakai H, Suzuki S, Mizuguchi T, et al. Rapid detection of gene mutations responsible for non-syndromic aortic aneurysm and dissection using two different methods: resequencing microarray technology and next-generation sequencing. Hum Genet. 2012;131:591–599.
Frank M, Albuisson J, Ranque B, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23:1657–1664.
Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16:881–888.
Buchan JG, Alvarado DM, Haller GE, et al. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum Mol Genet. 2014;23:5271–5282.
Acknowledgements
We are grateful to the patients. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (GR 3660/3–1 to G.R.) and by a grant from the BMBF (GeNeRARe, TP6 to G.R. and K.K., funding code 01GM1519E). All P/LP variants and associated phenotypes have been submitted to the ClinVar database (SUB4899534). The results summarized here are in part from the PhD thesis of S.R.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Renner, S., Schüler, H., Alawi, M. et al. Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients. Genet Med 21, 1832–1841 (2019). https://doi.org/10.1038/s41436-019-0435-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0435-z
Keywords
This article is cited by
-
Expanding the clinical spectrum of COL2A1 related disorders by a mass like phenotype
Scientific Reports (2022)
-
Role of Clinical Genetic Testing in the Management of Aortopathies
Current Cardiology Reports (2021)
-
Clinically relevant variants in a large cohort of Indian patients with Marfan syndrome and related disorders identified by next-generation sequencing
Scientific Reports (2021)
