
Abstract
Purpose
We have evaluated deficiencies in existing diagnostic criteria for neurofibromatosis 2 (NF2).
Methods
Two large databases of individuals fulfilling NF2 criteria (n = 1361) and those tested for NF2 variants with criteria short of diagnosis (n = 1416) were interrogated. We assessed the proportions meeting each diagnostic criterion with constitutional or mosaic NF2 variants and the positive predictive value (PPV) with regard to definite diagnosis.
Results
There was no evidence for usefulness of old criteria “glioma“ or “neurofibroma.” “Ependymoma” had 100% PPV and high levels of confirmed NF2 diagnosis (67.7%). Those with bilateral vestibular schwannoma (VS) alone aged ≥60 years had the lowest confirmation rate (6.6%) and reduced PPV (80%). Siblings as a first-degree relative, without an affected parent, had 0% PPV. All three individuals with unilateral VS and an affected sibling were proven not to have NF2. The biggest overlap was with LZTR1-associated schwannomatosis. In this category, seven individuals with unilateral VS plus ≥2 nondermal schwannomas reduced PPV to 67%.
Conclusions
The present study confirms important deficiencies in NF2 diagnostic criteria. The term “glioma” should be dropped and replaced by “ependymoma.” Similarly “neurofibroma” should be removed. Dropping “sibling” from first-degree relatives should be considered and testing of LZTR1 should be recommended for unilateral VS.
Similar content being viewed by others

INTRODUCTION
The two main sets of diagnostic criteria for neurofibromatosis 2 (NF2) date back to 1987 (ref. 1) and 1992 (ref. 2), although a points-based system was devised in 2011 (ref. 3). The Manchester criteria devised in 1992 (ref. 2) appear to be the most widely used and are superior to the 2002 criteria.4 Deficiencies were still noted because individuals with de novo NF2 often had a prolonged period with signs of NF2, but without meeting diagnostic criteria.4 More recently, several criteria have received more scrutiny. The term “glioma“ is increasingly seen as an incorrect descriptor. There is no convincing evidence that high-grade glioma is part of NF2 (ref. 5), with the great majority of NF2-associated spinal cord lesions being histologically proven to be ependymomas,6 and low-grade gliomas being relatively uncommon.5 The discovery of LZTR1-associated schwannomatosis in 2014 (ref. 7) led to the identification of substantial diagnostic overlap between schwannomatosis and NF2, particularly in those with unilateral vestibular schwannoma (VS) and multiple other nondermal schwannomas.8, 9 An update to the Manchester criteria was proposed to address this (Table 1) (ref. 9). Even the hallmark of NF2, bilateral VS, was recently shown to occur by chance. Indeed, calculations showed that in ~50% of those with symptomatic bilateral tumors alone >70 years, co-occurrence may have happened by chance.10 These deficiencies prompted us to reexamine the criteria using two large clinical and molecular databases.
MATERIALS AND METHODS
Two clinical databases curated since 1994 were utilized. A database of 1460 patients (1210 [83%] from residents in the UK) meeting existing NF2 diagnostic criteria, or harboring a constitutional pathogenic or likely pathogenic NF2 variant, or mosaic pathogenic variant classified as occurring at lower than 50% allele frequency or identified in two anatomically distinct NF2 related tumors. The main diagnostic criteria utilized were the Manchester criteria.2 As these are expanded National Institutes of Health (NIH) criteria1 all those meeting these original criteria are included. The Baser criteria3 are more restrictive, therefore all those meeting “definite” NF2 criteria were also included. A second database containing 1429 individuals who had undergone molecular analysis with one or more NF2 diagnostic criteria without fulfilling full clinical NF2 criteria was also interrogated. Thirteen individuals appear in both databases (9 mosaic for NF2 and 4 with a sporadic VS and a pathogenic NF2 variant). Seven patients with LZTR1 variants and a clinical NF2 diagnosis were in both databases but not included in the 1429. Thus 1416 additional patients were evaluated.
Ethical approval was obtained from the North West 7–Greater Manchester Central Research Ethics Committee (reference 10/H1008/74).
Each main diagnostic feature was taken as a major criterion. Thus bilateral VS, unilateral VS, multiple meningiomas, and an affected first-degree relative with NF2 were taken as the major criteria leading to diagnosis. Whichever major criterion was met first, ensuring a confirmed diagnosis (if required with sufficient minor criteria), was taken as the main route to diagnosis, e.g., an individual presenting with unilateral (VS) aged 25 years and a meningioma aged 27 years before developing a contralateral VS aged 30 years, only met diagnostic criteria at 30 years due to bilateral VS. If, however, two meningiomas were present aged 27 years, the “unilateral VS plus 2 other” category would have fulfilled diagnostic criteria at age 27 years. A diagnosis based on unilateral VS plus ≥2 meningiomas was included in the unilateral VS plus 2 other category rather than in the ≥2 meningiomas + unilateral VS category.
Separate analysis was carried out on those who had an ependymoma without bilateral VS at diagnosis and all those meeting the ≥2 meningioma category without bilateral VS at diagnosis (including the unilateral VS criterion). Late onset (age ≥60 years) of bilateral VS alone was assessed separately.
Differences in age at diagnosis were compared across groups (full constitutional, mosaic/presumed mosaic, and pathogenic variant not found) using the Kruskal–Wallis or the Mann–Whitney U test where appropriate. All p values were two-sided.
Molecular analysis
All individuals underwent lymphocyte DNA analysis for NF2, with additional analysis in LZTR1 and SMARCB1 in cases meeting the unilateral VS category and those with multiple schwannomas. NF2 genetic testing of lymphocyte DNA (and tumor when available) included exon sequencing and multiple ligation-dependent probe amplification (MLPA). In addition loss of heterozygosity (LOH) was assessed with intragenic polymorphic markers and flanking markers in tumors. Similar analysis was performed for LZTR1 and SMARCB1. Since 2013, clinical genetic testing has used next-generation sequencing (NGS). Individuals with de novo NF2 and learning problems also had chromosome analysis for ring 22 (n = 7-identified) and those with unfound familial NF2 were tested for translocations (n = 2-identified). Mosaicism was considered definite when either a pathogenic/likely pathogenic variant (class 4/5) was detectable in blood (often only after NGS guided by tumor analysis) or an identical pathogenic variant was found in two anatomically distinct tumors. A third category of probable mosaicism included individuals fulfilling NF2 diagnostic criteria, but with only one tumor available for analysis, with both mutational events found in one tumor and no variant detectable in blood. An NF2 diagnosis was refuted when molecular events were not consistent between two tumors, or a constitutional or mosaic NF2 variant was not identified and/or a pathogenic variant was found in another gene (e.g., LZTR1).
Positive predictive value (PPV) of diagnosis was calculated for those with definite molecularly confirmed or refuted NF2. Identification of an LZTR1 pathogenic variant in the absence of a germline NF2 variant, or the absence of a common genetic variant in two NF2-associated tumors, also excluded NF2.
RESULTS
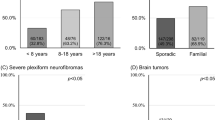
In total there were 1029 de novo individuals (3 with an affected sibling, but unaffected parents), and 332 with an affected parent and sufficient information to assess diagnostic category (total = 1361). The 1029 de novo cases were diagnosed at a median age of 34 years (range 0.5–86) whereas inherited cases were diagnosed at a median age of 22 years (range 0.2–82). Summary data of molecular analysis in those meeting existing criteria are presented in Table 2. The population was divided into de novo (clinical/molecular) cases (without a known affected parent) and those with an affected parent. Median age at diagnosis in each molecular category—constitutional pathogenic variant, mosaic/presumed mosaic variant, and no pathogenic variant found—is presented for de novo cases in Table 3.
De novo or with unaffected parent
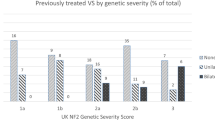
There were high levels of PPV for a definite confirmed diagnosis in all diagnostic categories (Table 2) except for individuals with unilateral VS plus a sibling with NF2, and individuals with unilateral VS plus ≥2 schwannomas. However, across all categories, there were low levels of NF2 confirmation (19.6–31.9%) in those initially presenting with unilateral VS. This is particularly concerning in the unilateral VS plus ≥2 schwannomas category where only 27.6% had a definite NF2 diagnosis and 7/58 (12%) were ultimately found to have a pathogenic LZTR1 variant. Of four cases without an NF2 or LZTR1 variant identifiable in blood, but for whom two tumors were available for analysis, three carried an identical NF2 variant in both tumors, while one had NF2 c.169C>T,p.(Arg57Ter) in one tumor; LOH 22q of the same allele in both tumors (including SMARCB1), and no LZTR1 or SMARCB1 variant in either tumor. This excludes an NF2 diagnosis in this last case. Therefore, along with the LZTR1 cases, of those given a definite diagnosis on clinical grounds, 8/24 (33%) did not have NF2 (we cannot exclude very low level mosaicism). All NF2-negative cases had LZTR1 and SMARCB1 analysis. This case with refuted NF2 received brain and mantle radiotherapy for lymphoreticular malignancy in late adolescence (case 157731, Table 4) and subsequently developed unilateral VS aged 40 with a C8 spinal lesion and axillary schwannoma. He subsequently developed a malignant peripheral nerve sheath tumor (MPNST) aged 51 in the C8 lesion, and thyroid cancer. Neither the C8 nor the axillary lesion had an identifiable NF2 variant or chromosomal loss, ruling out the c.1574+1G>A variant found in the VS as the causative variant. Five further cases with childhood radiotherapy are shown in Table 4. Case 9116 had a c.241-9A>G splicing variant confirmed after fractionated radiotherapy for bilateral optic pathway meningioma. Bilateral VS were identified 2 years later. The remainder developed NF2 tumors 12–25 years later consistent with radiation induced tumors.
The lowest detection rate for pathogenic NF2 variants was found in individuals with bilateral VS diagnosed aged ≥60 years. Overall, only 4/61 (6.6%) had a constitutional pathogenic variant (c.600-447_600-445delins8; c.19delT, p.[Ser7ProfsTer3]; c.15delC, p.[Ile5MetfsTer5]; c.1737+3A>T). There was no confirmed case of mosaicism in this category. Although three individuals had two pathogenic variants identified in one tumor, neither was detectable in blood. In a fourth case, two tumors were analyzed and different NF2 pathogenic variants were identified in each tumor, excluding an NF2 diagnosis.10
The median age at onset was significantly lower for each diagnostic category in individuals with a constitutional NF2 variant versus those with no variant identified. Mosaic cases were usually intermediate.
UVS in sibling or parent of NF2 case
Two individuals with unilateral VS were diagnosed after having a child diagnosed with NF2. Mosaic NF2 (allele frequency 10%) was diagnosed in one parent after a daughter was diagnosed with bilateral VS. A second parent was diagnosed with unilateral VS aged 22 and was only diagnosed with NF2 after his child developed bilateral VS in childhood. Cutaneous examination revealed likely schwannomas, but DNA confirmation was never undertaken. Three unrelated individuals with unilateral VS and a sibling with NF2 (parents unaffected) did not carry the pathogenic variant identified in their sibling. The VS were diagnosed aged 29, 39, and 49 years. Full NF2, LZTR1, and SMARCB1 analysis proved negative in blood. For the 29 year old the variant identified in tumor was not seen in blood. There were no situations in 1361 cases in which an individual with unilateral VS, an affected sibling, and unaffected parents would have been diagnosed with NF2.
Multiple meningiomas
Although a single meningioma is in the “other” category, ≥2 meningiomas is a “major” criterion. When analyzing ≥2 meningiomas separately in all categories including unilateral VS + 2 meningiomas, the detection rates were higher and more specific than the unilateral VS category. Overall, 52/137 (38%) with ≥2 meningiomas, had confirmed NF2 with no refuted diagnoses, compared with 45/207 (21.7%) with unilateral VS as major criterion (p = 0.001). For 32 individuals with ≥2 meningiomas the NF2 diagnosis was made with two additional NF2 features, but no VS, and 17/32 (53%) had molecularly confirmed NF2. This was particularly useful in childhood with 11/12 children diagnosed aged <15 years having a full constitutional pathogenic variant. Four cases were mosaic. A 60 year old with seven meningiomas and four spinal schwannomas had a c.169C>T p.(Arg57Ter) variant (4% lymphocyte allele frequency). Three further cases aged 47, 48, and 51 had an identical pathogenic variant found in two anatomically distinct meningiomas.
Five deceased unrelated parents with multiple meningiomas and no known VS had an affected child with NF2 and bilateral VS. Unfortunately, no material was available to confirm the constitutional pathogenic variant in the children. A sixth parent with six meningiomas (died aged 68 years) who had a deceased child with NF2, had no pathogenic variant identified in blood and no material was available from the daughter. Four of the six parents were males.
No SMARCB1 variant positive patients in the second Manchester database met NF2 criteria with ≥2 meningiomas (3/70 had a single meningioma). Fifty individuals with multiple meningiomas were tested, including 20 who meet NF2 criteria, and no SMARCB1 variants were found. Eight unrelated individuals with multiple meningiomas, but no other features of NF2, had germline pathogenic variants in SMARCE1 (refs. 11, 12). All eight individuals had clear cell meningiomas, rather than fibroblastic or transitional meningiomas, which are more common in NF2.
Intrinsic brain and spinal cord tumors
Most intrinsic tumors in NF2 were presumed ependymomas with very few undergoing resection. Of those intrinsic tumors with pathological confirmation, four were low-grade gliomas and only one was a high-grade glioma occurring after previous irradiation.5 None of these tumors would have aided an earlier diagnosis. There were 157 (12%) confirmed (n = 10) or presumed (n = 147) spinal cord ependymomas. The presence of an ependymoma increased the likelihood of identifying a pathogenic variant in those without bilateral VS to 68% (21/31), which was significantly higher than all other categories (p < 0.0001). The addition of ependymoma to the criteria would have advanced diagnosis by 1–23 years in 18 individuals who would not otherwise have met criteria. In three cases, an apparently sporadic ependymoma aged 13, 14, and 24 years would have led to an even earlier diagnosis by 2–4 years if genetic analysis of NF2 was initiated at time of ependymoma diagnosis.
Ocular features
The database did not hold extensive ocular features on NF2 patients. Nonetheless, undertaking molecular analysis on children with visual symptoms revealing retinal hamartoma or epiretinal membranes would have led to an earlier diagnosis before VS was diagnosed in at least 15 de novo children. In one child, the presence of amblyopia and epiretinal membranes led to mutational analysis that identified a pathogenic NF2 variant when the child was one year old (Table 2).
Neurofibroma
At least 67 (5%) NF2 patients had a pathology report stating “neurofibroma.” Most of these (n > 20) that underwent secondary pathology review were reclassified as schwannoma. Even assessing those without pathology review, none would have led to an earlier diagnosis of NF2 using existing criteria.
Offspring of NF2 affected individuals
No particular issues were identified to suggest deficiencies in the diagnostic criteria in this category, although a single case of unilateral VS aged 52 years in the daughter of a late-onset case with bilateral VS aged 75 years may reflect an inaccurate diagnosis. No pathogenic variant was identified in the woman or her mother. Overall, detection rates were >95% in keeping with detection rates for familial NF2.
DISCUSSION
The present study confirms a number of deficiencies with the 1987 NIH and1 1992 Manchester criteria.2 Perhaps the most pressing need is to change the term “glioma” to “ependymoma“ (Table 1). It is clear from radiological and pathological features that the predominant central nervous system (CNS) intrinsic tumor seen in NF2 is a spinal cord ependymoma.5, 6 These have generally been treated conservatively as they are normally indolent, but timely surgery clearly has a place in those with tumors over 15 mm in length.13 Ependymomas are very useful in classifying NF2 in those with unilateral VS9 or multiple meningiomas, providing the highest pathogenic variant detection rate (68%) of all de novo categories.
The main diagnostic overlap is with schwannomatosis. Here, we document two de novo cases with unilateral VS and ≥2 nondermal schwannomas (without other NF2 features), harboring LZTR1 pathogenic variants, bringing the total number to seven.9 Therefore, of those with a clinically confirmed diagnosis, only 67% have NF2, and the majority of these are mosaic. It is likely that the great remainder of those in this category without confirmed diagnosis have NF2 because they do not have an LZTR1 variant in blood or tumor. However, at least one further case without a SMARCB1 or LZTR1 variant had two tumors with divergent NF2 variants, excluding NF2 and potentially confirming a missed LZTR1 variant or another chromosome 22 schwannomatosis gene. Molecular analysis should be mandatory to confirm whether individuals with unilateral VS and other schwannomas have NF2 or LZTR1-related schwannomatosis because the consequences of these disorders, and differences in transmission risk, will be substantial, particularly because those with NF2 variants only detectable in tumors will have a very low transmission risk to children.14 Similarly, those with multiple nonvestibular schwannomas, without other NF2 features, may have mosaic NF2. In this report, 3 of 13 cases in this category have developed a VS and 10 further cases have not.15 About 50% of apparent schwannomatosis cases without an LZTR1 or SMARCB1 variant have mosaic NF2 (ref.15).
The present report also confirms that multiple meningiomas are as useful as, or better than, unilateral VS as a major criterion. The NF2 pathogenic variant detection rate is significantly higher in de novo cases meeting criteria without bilateral VS in those with ≥2 meningiomas than in those with unilateral VS. A number of parents of NF2 cases had multiple meningiomas and it is likely that these were mosaic for the pathogenic NF2 variant (the fact that 4/6 were male is unusual). Multiple meningiomas account for only 5% of patients with meningioma and at least 20% of these have NF2 (ref. 16). The population lifetime risk for multiple meningioma without NF2 is likely to be no more than 1 in 10–20,000 (ref. 16). Therefore, a chance association with NF2 appears less likely than unilateral VS, which occurs in 1 in 1000 people in their lifetime.10 The main diagnostic overlap concern for multiple meningiomas is with SMARCB1-associated schwannomatosis. Although we have not found SMARCB1 pathogenic variants in any case with ≥2 meningioma, occasional SMARCB1-associated multiple meningioma families have been described.17 For individuals with multiple meningiomas and no other features, it is possible that these may be caused by a germline SUFU variant (seen in a large Finnish family)18 or SMARCE1 (for clear cell meningiomas).11, 12
The present report also questions the use of a sibling with an unaffected parent as a diagnostic criterion. There are only two reports in the literature of siblings with NF2 and no affected parent.19, 20 Neither case presented with unilateral VS only. The likelihood of a sibling presenting only with unilateral VS is small because only around 5% of de novo NF2 patients present with an apparently sporadic unilateral VS.21 Nearly all of these are mosaic,21 whereas a sibling would have a full constitutional change. This scenario also depends on the parent having confined gonadal mosaicism, which appears extremely rare in NF2 because nearly all cases of parental mosaicism involve at least some level of detection in other tissues.14 All three cases in the present report who have unilateral VS and an NF2 affected sibling had not inherited the pathogenic variant identified in the sibling. Thus, the term “first-degree relative” should probably exclude siblings with unaffected parents, i.e., no evidence of deafness or NF2 tumors (Table 1), although molecular testing should clarify the situation in most instances.
Ophthalmic features consistent with NF2 in childhood should prompt molecular analysis.22, 23 A number of children could have had an earlier diagnosis with timely genetic assessment. Retinal hamartoma and childhood epiretinal membranes should be considered potential “minor” criteria for NF2 in addition to juvenile subcapsular and cortical cataracts.22,23,24
The neurofibroma has given neurofibromatosis its name. However, true pathological neurofibromas are rare in NF2 (ref. 25). Nonetheless, about 26% of nerve sheath tumors in NF2 have features of schwannoma and neurofibroma and are more correctly designated as hybrid tumors.25 It is likely that until this pathological term becomes universal that NF2 patients will continue to get an “inaccurate” diagnosis of neurofibroma. Such a diagnosis in a patient with NF2 features should prompt secondary pathology review. The continued use of “neurofibroma” within NF2 criteria is highly questionable (Table 1).
The final criterion that needs addressing is the hallmark of NF2 itself, bilateral VS. Both early criteria1, 2 make bilateral VS sufficient for diagnosis although the points system from 2011 includes an age cutoff of 30 years such that bilateral VS >30 years alone did not meet criteria for definite NF2. We have previously calculated that 1 in 2 million people will develop bilateral VS by chance10 and that ~50% of those with symptomatic bilateral VS ≥70 are due to chance. Increasing use of magnetic resonance imaging has shown that many people with bilateral VS identified in older life are asymptomatic. The very low detection rate for pathogenic variants of 6.6% (4/61) in isolated bilateral VS aged ≥60 is highly significantly less than in other diagnostic categories: 52/137 (38%) for ≥2 meningiomas (p < 0.0001) and 45/209 (21.5%) p = 0.0075 for unilateral VS plus two other. Nonetheless, the four identified with pathogenic variants had hypomorphic de novo variants (two exon 1 frameshift variants and two splicing variants) that still have important implications for children.26 If an age limit for definite NF2 were introduced for bilateral VS it would be vital to address offspring risks.
A final consideration is that schwannomas and meningiomas are radiation inducible tumors, especially with therapeutic radiation in childhood.27 In one study, among 3013 patients treated with radiotherapy before the age of 16, mostly for enlarged tonsils,28 70 (2.3%) developed neural tumors, with 7 developing multiple schwannomas or meningiomas. This is far higher than the birth incidence of NF2 and schwannomatosis combined.15 More recently, 3/33 sporadic adults meeting NF2 criteria in Israel had received cranial radiotherapy in childhood and none had an identifiable NF2 variant on blood analysis.29 We presented five further cases with childhood radiotherapy and no NF2 variant on blood analysis. In one of these cases the NF2 diagnosis could be refuted. As such, in individuals meeting NF2 criteria only through tumors arising >8 years post–childhood radiotherapy, their tumor should be considered more likely to be caused by radiation than NF2 (ref. 30).
The current study has some limitations. Ideally, to evaluate the true ability to provide a definite diagnosis of each criterion, two tumors from anyone without confirmed NF2 should be analyzed. In those with late-onset bilateral VS it is extremely rare to remove more than one and a high proportion of those who are treated receive radiation therapy. In 30 patients without LZTR1 variants that had two tumors analyzed, 3 (10%) refuted the NF2 diagnosis. As such, the values we report for the proportion of those with a definite diagnosis (PPV) are likely to be overestimates, particularly in categories with a low overall NF2 detection rate. Nonetheless, our study represents by far the largest assessment of diagnostic criteria based on nearly 3000 patients with molecular analysis. It includes potentially all identified NF2 cases in England through the four designated highly specialized commissioned centers15 and includes referrals for molecular testing of all those with a >1% chance of harboring an NF2 pathogenic variant in the last 8 years. Although molecular confirmation of NF2 appears very low, this is likely to be attributable to mosaicism, because most evaluable cases (90%) with two tumors had an identical NF2 variant detected in each tumor. The detection rate of 95% (sensitivity) for the second generation is reflected in Table 2 and our previous research.15 We did not evaluate cerebral calcification as the use of computed tomography (CT) scans has been limited since 1992. Because we would not recommend use of CT solely to identify calcification, particularly in childhood, we would not recommend retaining this criterion due to concerns over the ability to provide a definite diagnosis.
In conclusion, this report has identified a number of clear deficiencies in the current diagnostic criteria for NF2. There is a pressing need to develop new consensus criteria for NF2 that differentiate NF2 from schwannomatosis and remove criteria with poor ability to provide a definite diagnosis.
References
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578.
Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–618.
Baser ME, Friedman JM, Joe H, et al. Empirical development of improved diagnostic criteria for neurofibromatosis 2. Genet Med. 2011;13:576–581.
Baser ME, Friedman JM, Wallace AJ, Ramsden RT, Joe H, Evans DG. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59:1759–1765.
King AT, Rutherford SA, Hammerbeck-Ward C, et al. High-grade glioma is not a feature of neurofibromatosis type 2 in the unirradiated patient. Neurosurgery. 2018;83:193–196.
Hagel C, Stemmer-Rachamimov AO, Bornemann A, et al. Clinical presentation, immunohistochemistry and electron microscopy indicate neurofibromatosis type 2-associated gliomas to be spinal ependymomas. Neuropathology. 2012;32:611–616.
Piotrowski A, Xie J, Liu YF, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46:182–187.
Smith MJ, Isidor B, Beetz C, et al. Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology. 2015;84:141–147.
Smith MJ, Bowers NL, Bulman M, et al. Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1-related schwannomatosis. Neurology. 2017;88:87–92.
Evans DG, Freeman S, Gokhale C, et al. Bilateral vestibular schwannomas in older patients: NF2 or chance? J Med Genet. 2015;52:422–424.
Smith MJ, Wallace AJ, Bennett C, et al. Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol. 2014;234:436–440.
Smith MJ, O’Sullivan J, Bhaskar SS, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet. 2013;45:295–298.
Kalamarides M, Essayed W, Lejeune JP, et al. Spinal ependymomas in NF2: a surgical disease? J Neurooncol. 2018;136:605–611.
Evans DG, Wallace A. An update on age related mosaic and offspring risk in neurofibromatosis 2 (NF2). J Med Genet. 2009;46:792.
Evans DG, Bowers NL, Tobi S, et al. Schwannomatosis: a genetic and epidemiological study. J Neurol Neurosurg Psychiatry. 2018;89:1215–1219.
Antinheimo J, Sankila R, Carpen O, Pukkala E, Sainio M, Jaaskelainen J. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54:71–76.
van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2011;13:1–7.
Aavikko M, Li SP, Saarinen S, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet. 2012;91:520–526.
Sestini R, Vivarelli R, Balestri P, Ammannati F, Montali E, Papi L. Neurofibromatosis type 2 attributable to gonosomal mosaicism in a clinically normal mother, and identification of seven novel mutations in the NF2 gene. Human Genet. 2000;107:366–371.
Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450–461.
Evans DG, Raymond FL, Barwell JG, Halliday D. Genetic testing and screening of individuals at risk of NF2. Clin Genet. 2012;82:416–424.
Ruggieri M, Pratico AD, Serra A, et al. Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies. Acta Otorhinolaryngol Ital. 2016;36:345–367.
Anand G, Vasallo G, Spanou M, et al. Diagnosis of sporadic neurofibromatosis type 2 in the paediatric population. Arch Dis Child. 2018;103:463–469.
Painter SL, Sipkova Z, Emmanouil B, Halliday D, Parry A, Elston JS. Neurofibromatosis type 2-related eye disease correlated with genetic severity type. J Neuroophthalmol. 2018. https://doi.org/10.1097/WNO.0000000000000675. [Epub ahead of print].
Montgomery BK, Alimchandani M, Mehta GU, et al. Tumors displaying hybrid schwannoma and neurofibroma features in patients with neurofibromatosis type 2. Clin Neuropathol. 2016;35:78–83.
Hexter A, Jones A, Joe H, et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: a UK national analysis of 1192 patients. J Med Genet. 2015;52:699–705.
Ron E, Modan B, Boice JD Jr, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988;319:1033–1039.
Sznajder L, Abrahams C, Parry DM, Gierlowski TC, Shore-Freedman E, Schneider AB. Multiple schwannomas and meningiomas associated with irradiation in childhood. Arch Intern Med. 1996;156:1873–1878.
Bokstein F, Dubov T, Toledano-Alhadef H, et al. Cranial irradiation in childhood mimicking neurofibromatosis type II. Am J Med Genet. 2017;173:1635–1639.
Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet. 2006;43:289–294.
Acknowledgements
The authors wish to acknowledge National Health Service (NHS) England for their support of the National NF2 program. D.G.E., E.F.H., and M.J.S. are supported by the Manchester National Institute for Health Research (NIHR) Biomedical Research Centre (IS-BRC-1215-20007)
English Specialist NF2 Research Group members:
Cambridge/Central: Patrick Axon, Juliette Gair, James Tysome, Neil Donnelly, Lucy Raymond, Anke Hensiek, Rajesh Jena, Robert Macfarlane, Richard Mannion, James Nicholson, Brinda Muthusamy, Amy Taylor, Richard Price, Gabriella Rands, Nicola Gamazo, Zebunnisa Daniel Scoffings, Sarah Jefferies, Richard Knight, Tamara Lamb, Vanat, Yu Chuen Tam, Karen Foweraker, Fiona Harris, David Heney, Paul Sanghera, Richard Irving, Peter Monksfield, Saba Sharif, Nicola Ragge, Carolyn Smyth, Julian Barwell, Martin English. London: Shazia Afridi, Rosalie Ferner, Rupert Obholzer, Victoria Williams, Chris Hammond, Karine Lascelles, Chris Skilbeck, Shakeel Saeed, Adam Shaw, Angela Swampillai, Suki Thomson, Nick Thomas, Eleni Maratos, Sinan Barazi, Rebecca Mullin, Susie Henley, Sally Trump, Vanessa Everett, Terry Nunn, Charles Nduka. Manchester/North: D Gareth Evans, Raji Anup, Chris Duff, Simon R Freeman, Emma Stapleton, Nicola Jarvis, Ian Kamaly-Asl, Andrew King, Mark Kellett, John-Paul Kilday, Simon Lloyd, Connor Malluci, Deborah Mawman, Catherine McBain, Roger Laitt, Martin O’Driscoll, Martin McCabe, Mary Perry, Scott Rutherford, Kirsty Henshaw, Stavros Stivaros, Owen Thomas, Grace Vassallo, Charlotte Hammerbeck-Ward, Omar Pathmanaban, Jincy Kurian. Oxford/South-West: Claire Blesing, Kate Browne, Rosie Crabtree, Lucy Cogswell, Louise Dalton, Caroline Dodridge, Beatrice Emmanouil, Henk Giele, Dorothy Halliday, C Oliver Hanemann, Wendy Howard, Sanjeeva Jeyaretna, Richard Kerr, Elle Mace, Sam MacKeith, Anne May, Allyson Parry, Peter Pretorius, James Ramsden, Carolyn Redman, Srilakshmi Sharma Ros Taylor, Helen Tomkins, Shaun Wilson, Rachael Woolrich.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Evans, D.G., King, A.T., Bowers, N.L. et al. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet Med 21, 1525–1533 (2019). https://doi.org/10.1038/s41436-018-0384-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0384-y
Keywords
This article is cited by
-
The genetic landscape and possible therapeutics of neurofibromatosis type 2
Cancer Cell International (2023)
-
Correlation between genotype and phenotype with special attention to hearing in 14 Japanese cases of NF2-related schwannomatosis
Scientific Reports (2023)
-
Prevalence and natural history of schwannomas in neurofibromatosis type 2 (NF2): the influence of pathogenic variants
European Journal of Human Genetics (2022)
-
ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis
European Journal of Human Genetics (2022)
-
Managing Headache Disorders Associated with Tuberous Sclerosis and Neurofibromatosis
Current Pain and Headache Reports (2022)
