
Abstract
Purpose
Wolf–Hirschhorn syndrome (WHS) is a genomic disorder with a recognizable dysmorphology profile caused by hemizygosity at 4p16.3. Previous attempts have failed to map the minimal critical locus to a single gene, leaving open the possibility that the core phenotypic components of the syndrome are caused by the combined haploinsufficiency of multiple genes.
Methods
Clinical exome sequencing and “reverse” phenotyping.
Results
We identified two patients with de novo truncating variants in WHSC1, which maps to the WHS critical locus. The phenotype of these two individuals is consistent with WHS, which suggests that haploinsufficiency of WHSC1 is sufficient to recapitulate the core phenotype (characteristic facies, and growth and developmental delay) of this classic microdeletion syndrome.
Conclusion
Our study expands the list of microdeletion syndromes that are solved at the single-gene level, and establishes WHSC1 as a disease gene in humans. Given the severe nature of the reported variants, the full phenotypic expression of WHSC1 may be further expanded by future reports of milder variants.
Similar content being viewed by others


Introduction
In 1965 a novel multiple congenital anomalies/mental retardation (MCA/MR) syndrome caused by partial 4p deletion was described.1 Wolf–Hirschhorn syndrome (WHS) (OMIM 194190) is now a well-recognized neurodevelopmental and craniofacial malformation syndrome caused by variably sized deletions of chromosomal region 4p16.3. The core WHS phenotype is defined by a typical craniofacial appearance, growth delay, mild-to-profound developmental delay, and seizures (or EEG anomalies).2 Characteristic craniofacial features include microcephaly, prominent glabella, widely spaced and prominent eyes, a “Greek warrior helmet appearance” of the nose, a broad nasal tip, a short philtrum, and downturned corners of the mouth. The frequency of WHS is estimated to be 1 in 50,000 to 1 in 20,000 births, and is more frequent in females than in males (2:1).2,3
As with many microdeletion syndromes, early studies to narrow the WHS critical region (WHSCR) relied on karyotypic deletion mapping, which successfully linked the core phenotype to a 165-kb locus on 4p16.3.4 WHSCR contained two genes and these were referred to as WHS candidate gene 1 (WHSC1, contained only partly within WHSCR) and WHS candidate gene 2 (WHSC2, entirely contained within WHSCR). A recent study employing high-resolution molecular karyotyping (microarrays) narrowed WHSCR further to 109 kb that spans, in addition to WHSC1, two other genes, and the authors proposed WHSC1 as the most compelling candidate.5 However, because no patients with deletions of a single gene were reported, it remains unknown whether WHSC is a complex phenotype caused by haploinsufficiency of multiple genes or a single-gene disease with potential phenotypic modification caused by haploinsufficiency of the surrounding genes.
In a recent large-scale sequencing study of nearly 1,000 Saudi families with various clinical indications, we encountered a likely deleterious WHSC1 variant in a child with dysmorphic features, and growth and developmental delay.6 Here, we show upon careful review of the phenotype and through parental testing that this child’s phenotype is consistent with WHS and that the variant has arisen de novo. In addition, we were able to identify another child with WHS phenotype who also has a different de novo truncating variant in the same gene.
Subjects and methods
Both patients were evaluated on clinical basis. Clinical exome sequencing and Sanger validation were performed as described previously with proper informed consent.6 Additional consent to publish clinical photographs was also obtained.
Results
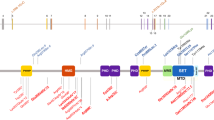
Patient 1: This 5-year-old girl, the only child of healthy unrelated parents, was referred to Clinical Genetics for evaluation of growth and developmental delay. Pregnancy was notable for intrauterine growth retardation necessitating induction of labor at 34 weeks of gestation (birth weight 1.5 kg) with subsequent admission to the neonatal intensive care unit (NICU) for weight management. Floppiness and developmental delay were noted in infancy. She only walked after her second birthday and spoke her first sentences at age 5 years. Growth delay persisted (current weight is 15.5 kg [−2.4 SD], height 97.5 cm [−2.8 SD] and OFC (occipitofrontal circumference) 47.5 cm [−2SD]), and she developed multiple aspiration pneumonia episodes. There was no history of seizures. Details of her physical examination are shown in Table 1 and Fig. 1. Abdominal ultrasound and skeletal survey were normal. Molecular karyotyping was normal. Clinical exome sequencing (WES) revealed the following variant in WHSC1 as the likely cause: NM_001042424:c.2518+1G>A, which was confirmed to be de novo (Fig. 1).

(a,b) Clinical photographs of patient 1 showing facial asymmetry, short philtrum, micrognathia, high forehead, prominent ears, downturned corners of mouth, and micrognathia. (c,d) Clinical photographs of patient 2 as (c) a young infant and (d) currently showing milder facial features compared to patient 1, including high forehead, broad nasal tip, and pointed chin. (e,f) Sequence chromatograms of the two de novo variants in WHSC1 with their locations indicated by colored asterisks. (g) WHSC1 protein cartoon. Note the location of the two truncating variants (denoted by colored asterisks) upstream of the critical SET domain
Patient 2: A 34-month-old boy with developmental and growth delay. He is the second child in a family of healthy consanguineous parents. His pregnancy was complicated by intrauterine growth retardation. He was born naturally at term with a birth weight of 2.3 kg. He was initially evaluated for growth and global developmental delay and was found to have hearing loss necessitating cochlear implant. Growth delay was progressive: at age 8 months, his weight was 6 kg (−3.8 SD), length 66 cm (−2 SD) and OFC 44 cm (28th centile), while at age 2 years, his weight was 8.8 kg (−4.1 SD), length 78 cm (−3.6 SD), and OFC 45.5 cm (−2 SD). Currently, he just started to pull to standing, he can use a spoon clumsily to eat, has stranger anxiety, only plays with his older brother, only understands basic commands, and has a limited single-word vocabulary of 10 simple words. Details of his physical examination are shown in Table 1 and Fig. 1. Brain magnetic resonance image (MRI), skeletal survey, and abdominal ultrasound were normal. Molecular karyotyping was normal. Clinical WES revealed the following variant in WHSC1 as the likely cause: NM_001042424:c.2803C>T:p.(R935*), which was confirmed to be de novo (Fig. 1). It also revealed a homozygous loss of function variant in MYO7A (NM_000260:exon17:c.2005C>T:p.(R669*)) as a likely explanation of deafness.
Discussion
Microdeletion syndromes are genomic disorders in which the phenotype-related deletions, typically too small for detection by karyotype, span more than one gene. They usually arise de novo and tend to have recurrent breakpoints due to the presence of flanking low-copy repeat gene clusters that predispose to genomic instability. Classical microdeletion syndromes include, amongst others, Angelman syndrome (15q11.2-q13), Prader–Willi syndrome (15q11.2-q13), Williams–Beuren syndrome (7q11.23), Smith–Magenis (17p11.2), velocardiofacial/DiGeorge syndrome (22q11.2), and WHS (4p16.3). Mapping of overlapping but distinct microdeletions has led to the delineation of critical intervals, and in some cases, to the discovery of a single causative gene. More recently, large scale WES has revealed deleterious variants in single genes that appear to recapitulate the phenotype of microdeletion syndromes, e.g., SETD5 and 3p25 microdeletion,7 WDR26 and 1q41q42 microdeletion,8 PUF60 and 8q24.3,9 and ANKRD11 and 16q24.3 microdeletion.10
Surprisingly, none of the previously published large-scale WES in outbred populations have identified deleterious variants in WHSC1, a gene previously linked to the critical region of WHS, and yet our WES analysis of less than 1,000 families from the highly consanguineous population of Saudi Arabia revealed two de novo variants in this gene.6 This may represent a chance occurrence or, alternatively, a higher propensity of our analytical pipeline to highlight likely deleterious variants in genes that are highly intolerant to haploinsufficiency in the human genome (WHSC1 has a pLI of 1.00).
Wolf–Hirschhorn syndrome candidate 1 (WHSC1), also known as multiple myeloma SET protein (MMSET) or nuclear receptor-binding SET domain-protein 2 (NSD2), is a SET domain histone methyltransferase, responsible for the methylation of H3K36. Histone modification is increasingly recognized as an important biological process in normal brain development as evidenced by the growing list of dominant and recessive intellectual disability genes that encode components of histone modifications.11,12,13 Some of the more commonly mutated histone modification genes in the context of intellectual disability include KMT2D, KDM6A, KDM1A, KMT2B, KMT2C, and KMT2A. The two variants we describe predict truncation upstream of the SET domain, which is necessary for the methylation of lysine-9 in the histone H3 N terminus, although the exact consequence of the splicing variant is harder to predict (Fig. 1).14
Prior to the advent of molecular karyotyping, only patients with suspected WHS would undergo specific testing for 4p16.3 microdeletion using multiplex ligation-dependent probe amplification (MLPA) or fluorescence in situ hybridization (FISH) leading to a clear bias in defining the core phenotype of the syndrome, which is typically defined as the classical dysmorphology profile, growth and developmental delay, and epilepsy. Unbiased genomewide view of individuals with various indications was made possible by molecular karyotyping, which as expected, revealed a much wider phenotypic spectrum than previously suspected. This is best appreciated in the large series by Baylor Miraca Genetics Laboratories, which reported 156 unrelated patients with copy-number variants (CNVs) involving 4p16.3 from over 60,000 patients studied.15 CNVs were detected prenatally in 8 patients, while the remaining 148 patients were analyzed postnatally. Among the postnatal patients, the primary indications for the chromosomal microarray study were developmental delay, dysmorphic features, multiple congenital anomalies, and seizures. Although detailed descriptions of the patient phenotype were not provided, it was clear that most patients were not suspected clinically to have WHS, which suggests that their phenotype may not have been typical.15
We argue that the two patients we report in this study have WHS caused by their de novo truncating variants in WHSC1. Given the very small number of our cohort, it is premature to conclude that lack of seizures necessarily indicates that WHSC1-related WHS phenotype is incomplete and that epilepsy is caused by other genes as suggested by others,16 especially because not all patients with classical 4p16.3 deletion have epilepsy.2 We also note that we lack EEG on the two patients so we cannot rule out subclinical epilepsy. Our finding that WHSC1 heterozygous loss of function recapitulates WHS makes it now possible to observe the full phenotypic spectrum of WHSC1-related phenotypes especially in the context of less severe variants, which may not necessarily be de novo in nature if the resulting phenotype is mild.
References
Hirschhorn K, Cooper HL, Firschein IL. Deletion of short arms of chromosome 4–5 in a child with defects of midline fusion. Humangenetik. 1965;1:479–82.
Battaglia A, Carey JC, South ST Wolf-Hirschhorn Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: a review and update. Am J Med Genet C Semin Med Genet. 2015;169:216–23.
Wright TJ, Ricke DO, Denison K, et al. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum Mol Genet. 1997;6:317–24.
Okamoto N, Ohmachi K, Shimada S, Shimojima K, Yamamoto T. 109 kb deletion of chromosome 4p16.3 in a patient with mild phenotype of Wolf-Hirschhorn syndrome. Am J Med Genet A. 2013;161A:1465–9.
Monies D, Abouelhoda M, AlSayed M, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–939
Grozeva D, Carss K, Spasic-Boskovic O, et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am J Human Genet. 2014;94:618–24.
Skraban CM, Wells CF, Markose P, et al. WDR26 haploinsufficiency causes a recognizable syndrome of intellectual disability, seizures, abnormal gait, and distinctive facial features. Am J Human Genet. 2017;101:139–48.
Dauber A, Golzio C, Guenot C, et al. SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24. 3 copy-number variant. Am J Human Genet. 2013;93:798–811.
Sirmaci A, Spiliopoulos M, Brancati F, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Human Genet. 2011;89:289–94.
Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–61.
Anazi S, Maddirevula S, Faqeih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2016;22:615–24.
Anazi S, Maddirevula S, Salpietro V, et al. Expanding the genetic heterogeneity of intellectual disability. Hum Genet. 2017;136:1419–29.
Nimura K, Ura K, Shiratori H, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–91.
Bi W, Cheung SW, Breman AM, Bacino CA. 4p16.3 microdeletions and microduplications detected by chromosomal microarray analysis: new insights into mechanisms and critical regions. Am J Med Genet A. 2016;170:2540–50.
Ho KS, South ST, Lortz A, et al. Chromosomal microarray testing identifies a 4p terminal region associated with seizures in Wolf–Hirschhorn syndrome. J Med Genet. 2016;53:256–63.
Acknowledgements
We thank the study families for their enthusiastic participation. We thank the team at the Medical Diagnostic Laboratory for their technical help. We acknowledge the support of the Saudi Human Genome Program, and King Salman Center for Disability Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Derar, N., Al-Hassnan, Z.N., Al-Owain, M. et al. De novo truncating variants in WHSC1 recapitulate the Wolf–Hirschhorn (4p16.3 microdeletion) syndrome phenotype. Genet Med 21, 185–188 (2019). https://doi.org/10.1038/s41436-018-0014-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0014-8
Keywords
This article is cited by
-
Auricular fistula: a review of its clinical manifestations, genetics, and treatments
Journal of Molecular Medicine (2023)
-
From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: a shifting paradigm through the description of a new case and a review of the literature
Italian Journal of Pediatrics (2022)
-
The first familial NSD2 cases with a novel variant in a Chinese father and daughter with atypical WHS facial features and a 7.5-year follow-up of growth hormone therapy
BMC Medical Genomics (2020)
-
De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype
BMC Medical Genetics (2019)
