
Abstract
We report the clinical, biochemical and genetic findings from a Spanish girl of Caucasian origin who presented with macrocephaly, dysmorphic facial features, developmental delay, hypotonia, combined oxidative phosphorylation (OxPhos) deficiency, epilepsy and anti-phospholipid antibodies (aPL). Whole-exome sequencing (WES) uncovered a heterozygous variant in the MTOR gene (NM_004958.3: c.7235A>T: p.(Asp2412Val)) that encodes for the Serine/threonine-protein kinase mTOR. The substrates phosphorylation experiments demonstrated that this variant exerts its effect by gain-of-function (GOF) and autosomal dominant mechanism. GOF variants in this protein have been associated with Smith-Kingsmore syndrome (SKS), a rare autosomal dominant disorder characterized by intellectual disability, macrocephaly, seizure, developmental delay and dysmorphic facial features. Furthermore, the mTOR pathway has been demonstrated previously to be involved in many types of endothelium injuries including the antiphospholipid syndrome (APS), a systemic autoimmune disease characterized by the production of aPL with recurrent vascular thrombosis. Therefore, our patient is the first one with an mTOR variant and diagnosed with SKS and APS. In conclusion, our data expand both the genetic and phenotypic spectrum associated with MTOR gene variants.
Similar content being viewed by others

Introduction
The MTOR gene (Mechanistic target of rapamycin kinase [MIM#601231]) encodes the Serine/threonine-protein kinase mTOR (P42345) [1,2,3] that belongs to the phosphatidylinositol 3-kinase (PI3K)-related kinase family. The protein is a key regulator of the pathway that senses and integrates a variety of nutrient-sensitive signals to regulate cellular metabolism, coordinating protein synthesis and mitochondrial activity to drive growth and proliferation [4]. The mTOR is the core component of two functionally distinct signaling complexes named mTOR complex 1 (mTORC1) and 2 (mTORC2). The primary effector for the nutrient-sensitive functions is mTORC1, while mTORC2 is implicated in cytoskeletal reorganization and cell survival [3].
Deregulation of the mTOR pathway has been implicated in a diverse range of human pathologies, including cancer, autoimmunity, cardiovascular diseases, neurodegenerative diseases and metabolic disorders [4]. Smith-Kingsmore syndrome (SKS; MIM#616638) [5] is a rare disorder caused by variants in the MTOR gene. The most consistent findings in SKS are intellectual disability (ID), developmental delay (DD), megalencephaly and seizures, and to date, only 10 variants in MTOR gene have been described in 27 families with SKS [6].
Antiphospholipid syndrome (APS; MIM#107320) is a systemic autoimmune disorder characterized by arterial or venous thrombosis, frequently accompanied by a moderate thrombocytopenia in the presence of antiphospholipid antibodies (aPL) [7]. The aPL includes lupus anticoagulant (LA), anti-cardiolipin antibodies (aCL), and anti-β2-glycoprotein-I antibodies (anti-β2GPI) [8]. Several studies have suggested that the mTOR pathway is involved in the vascular lesions associated with the APS [9], and that oxidative stress and mitochondrial dysfunction play also an important role in APS [10].
Herein, we describe a patient presenting SKS and APS type I, in whom we identified the de novo variant p.(Asp2412Val) in the MTOR gene by WES.
Materials and methods
Patient
The Ethic Committee of the Instituto de Investigación Hospital 12 de Octubre (i+12) approved the study, and written informed consent was obtained from the patient’s parents.
The Spanish patient is a girl of non-consanguineous healthy parents with a negative family history for congenital anomalies and ID. Her older brother is healthy, and her mother had a previous miscarriage of unknown cause at 8th week (Fig. 1a). At the 32 weeks of gestation, macrocephaly was detected by ultrasonography. The girl was born at 36 weeks of gestation by vaginal delivery, with a weight of 3.52 kg (+1.6SD), a body length of 50.0 cm (+1.0SD) and head circumference (HC) of 38.7 cm (+3.6SD) confirming the macrocephaly. Apgar scores were 9 and 10 after 1 and 5 min, respectively. Global DD was evident in the first months, when physical examination showed generalized muscular hypotonia and dysmorphic features consistent in macrocephaly, prominent high forehead, downslanting palpebral fissures, depressed nasal bridge, protuberant abdomen and umbilical hernia. Social smile, head control, sitting position, and bubbling appeared at 6, 9, 16, and 20 months of age respectively. At 2 years, tonsillectomy, adenoidectomy and tympanic drainage were performed due to upper respiratory tract infections. A metabolic screen, karyotype and array-CGH were normal. Magnetic resonance imaging (MRI) of the brain showed hypoplasia of the splenium (Fig. 2a) and a cavum vergae as normal variant (Fig. 2b). Cerebrospinal fluid analysis, including lactate, folic acid and neurotransmitters, was normal. Muscle biopsy showed a moderate atrophy of type IIb fibers in histological study without histochemical abnormalities. Mitochondrial respiratory chain (MRC) analysis in skeletal muscle showed deficiencies of complex I, III and IV, while complex II and citrate synthase activities were in the normal range (Table 1) [11]. Mitochondrial DNA (mtDNA) studies showed absence of deletions and depletion, and the whole mtDNA sequencing did not show any pathological variant. At 4 years of age she was able to walk, to run and to climb stairs and to communicate using gestures, vocal sounds and behavior. By the age of 8 years, she presented her first seizure. Levetiracetam was ineffective to treat epilepsy whereas with carbamazepine the patient became seizure-free. Cerebral MRI performed at 9 years old, demonstrated bilateral lesions in cortical and sub-cortical areas suggestive of chronic infarcts of small vessels and enlargement of the sulci because cerebral atrophy (Fig. 2c–e). The epilepsy event was accompanied by the appearance of frequent subcutaneous hematomas after minor injuries, reason why a complete hematological assessment was performed. Hematologic study demonstrated persistently thrombocytopenia, and positive anti-phospholipid antibodies (aPL): lupus anticoagulant (LA), anti-cardiolipin (aCL) and anti-β2-glycoprotein I antibodies. Then she was diagnosed with antiphospholipid syndrome (APS) type I according with the last Sydney criteria [8], and treatment with low dose acetylsalicylic acid was started. At 13 years old, and despite treatment compliance during 4 years, new lesions with the same characteristic appeared at MRI follow-up (Fig. 2f–i). Except for epilepsy, she did not present any changes on neurologic exam or any appreciable signs of neurological regression. She has been attending since the first years of life a school for children with special needs with slow but progressive neurodevelopment. However, she never learnt to speak or to be independent in daily living activities.

Genetic analysis. a Family pedigree showing the genotype of the de novo c.7235A>T variant in MTOR [NM_004958.3] that produces the change of aspartic acid (Asp/D) to valine (Val/V) in the position 2412 of the α isoform (p.(Asp2412Val)/p.(D2412V)) or 569 of the β isoform (p.(Asp569Val)/p.(D569V)). b Electropherograms showing Sanger sequence validation of the MTOR c.7235A>T (p.(D2412V)) variant. c Multiple sequence alignment of Serine/threonine-protein kinase mTOR (protein encoded by MTOR gene) region surrounding the p.D2412 (red) variant site in various species. d Schematic representation of human Serine/threonine-protein kinase mTOR splicing isoforms showing the position of p.(D2412V) (red, up) variant in the isoform α, and p.(D569V) (red, down) in the isoform β. The structural features are shown: HEAT repeats (Huntingtin, EF3, PP2A and TOR1), FAT (FRAP/TOR-ATM-TRRAP), FRB (FKBP12-Rapamycin-Binding), and KD (Kinase Domain)

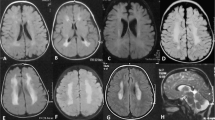
Magnetic resonance imaging (MRI). MRI studies in the follow up of the patient at the age of 2 (a, b), 9 (c, d, e) and 13 (f, g, h, i) years old. Sagittal T1-weighted image showed corpus callosum hypoplasia (arrow) (a), and axial Flair (Fluid attenuation inversion recovery) image showed cavum vergae (arrow) (b). Axial T2-weighted image at the basal ganglia showed enlargement of the extra-axial space of the left cerebral hemisphere (arrow) (c), and at the centrum semiovale showed lacuna porencephalic cysts (arrow) (d). Coronal Flair showed hyperintensity of the cortex and centrum semiovale in frontal left lobe (arrow) (e). Axial T2-weighted images (f, h) and axial SWI (Susceptibility weighted imaging) images (g, i) showed cystic like lesion in the head of the caudate (arrow, f) surrounded with hemosiderin (arrow, g), and lacunar porencephalic lesions in white matter of subcortical areas (arrow, h) with hemosiderin deposit (arrow, i)
Currently, at age of 13 years old, her clinical condition remains stable and her growth and weight gain are in the normal range. Thus, her weight is 48.4 kg (−0.2 SD), her length 147 cm (−1.6 SD) and her head circumference 61 cm (+5.27 SD). She has never presented venous thrombosis.
Genetic analysis
Whole-exome sequencing (WES) was performed (BGI-Hong Kong Co., Ltd) on genomic DNA obtained from patient, brother and parents blood, following a standard protocol previously described [12]. Briefly, the amplified DNA fragments were hybridized to the Agilent SureSelect Human All Exon V4 (51 Mb), the captured library was sequenced on a HiSeq 2000 platform, and the reads were aligned against the human reference genome (GRCh37 at UCSC) in order to obtain candidate variants. Nuclear variants and indels were prioritized according to the following criteria [13]: (i) variants that were rare in healthy individuals (allele frequency below 0.01) or new (not described within public databases); (ii) variants predicted to modify protein function (nonsense, splice site, coding indel, or missense variants); (iii) variants consistent with a recessive model of pathogenesis (including homozygous variants or two heterozygous variants present in the same gene) or (iv) variants consistent with a de novo dominant model of pathogenesis (variants new o with allele frequency below 10−5). Additional indications to prioritize the candidate genes were obtained by using predictive software scoring the likelihood for pathogenicity SIFT, Polyphen-2, MutPred and Variant Taster. Sanger sequencing of the patient, her brother and her parents was performed for candidate gene variant.
The data of the study (variant and phenotype) has been submitted to LOVD database (www.LOVD.nl/MTOR; patient ID 00218046).
Western-blot
The lymphocytes from control, parents, healthy brother and patient were lysed and the protein concentrations were determined using “Micro BCA Protein Assay Kit” (Thermo Fisher Scientific). Proteins were separated by a gradient 4–20% (w/v) SDS-PAGE, transferred to PVDF membrane (Bio-Rad) and then probed with antibodies against the different proteins. A goat anti-mouse or anti-rabbit was used as a secondary antibody. The western-blot signals were quantified by densitometry using NIH Image J version 1.49 software, and the intensities were normalized with that of β-actin. The “Relative expression” was calculated as ratios to the normal control values:
Antibodies
Antibodies included anti-mTOR (CST, 2983), anti-mTOR (phospho-Ser2448) (CST, 5536), anti-4E-BP1 (phospho-Thr37/46) (CST, 2855), anti-P70S6K (phospho-Ser371) (CST, 9208), anti-P70S6K (phospho-Thr389) (Abcam), anti-AKT (phospho-Ser473) (CST, 4060), anti-ULK1 (phosphor-Ser757) (CST, 6888), MitoBiogenesis western blotting cocktail (ab123545, Abcam, containing anti-SDHA, anti-MT-CO1 and β-actin), Total OXPHOS human WB antibody cocktail (ab110411, Abcam, containing anti-ATP5A, anti-UQCRC2, anti-SDHB, anti-MT-CO2 and anti-NDUFB8), anti-COX5A antibody (ab110262, Abcam), GUF1 polyclonal antibody (PA5-48469) (ThermoFisher Scientific), β-actin (Abnova), Goat anti-Rabbit IgG (H+L) secondary antibody, HRP conjugate (31460) and Goat anti-Mouse IgG (H+L) secondary antibody, HRP conjugate (31430) (ThermoFisher Scientific).
Statistical analysis
The data were analyzed using independent samples t test, and P values lower than 0.05 were considered statistically significant.
Results
Brain MRI findings
Regarding brain MRI findings, a corpus callosum shorter than normal (hypoplasia of the splenium) (Fig. 2a), a cavum vergae as a normal variant (Fig. 2b) and craniofacial malformation, were showed at the age of 2 years. MRI performed at the age of 9 years showed an enlargement of the extra-axial space of the left cerebral hemisphere due to cerebral atrophy (Fig. 2c). Also there were small porencephalic cysts with abnormal border signal, and diffuse white matter signal in the semioval centrum (Fig. 2d) and in several segments of the cortex (Fig. 2e). However, ischemic acute lesions were not considered since not tissue with restriction on diffusion weighted images were found. For this reason, all these radiological findings suggested that the lesions were chronic lesions caused presumably by some type of ischemic event. Finally, a new MRI acquired at 13 years old showed a progression of the disease. Thus, new lesions with hemosiderin deposit were found with SWI (Susceptibility weighted imaging) sequences: one affecting the left head of the caudate nucleus (Fig. 2f, g) and two in the white matter of semioval center (Fig. 2h, i).
Exome sequencing
The aforementioned analytic pipeline for nuclear variants and indels analysis was used to prioritize variants in genes that were rare and were predicted to be deleterious. We found a heterozygous missense variant (NM_004958.3: c.7235A>T:p.(Asp2412Val)/p.(D2412V) in exon 53 as numbered in NG_033239.1) in the MTOR gene (Mechanistic Target of Rapamycin Kinase) that encodes for the Serine/threonine-protein kinase mTOR (UniProt: P42345). Sanger sequencing of patient, her brother and her parents was performed, and the NC_000001.10:g.11174440T>A (chr1, hg19) variant was found to be de novo (Fig. 1b). The amino acid change is ascribed to “affects function” by using well-established in-silico tools of pathogenesis prediction (SIFT, PolyPhen2, MutPrep and Mutation Taster) (data not shown), and it is highly evolutionarily conserved (with PhastCons score of 1) from Homo sapiens to Xenopus tropicalis (Fig. 1c).
Functional effects
To evaluate the functional effect of the identified variant, we assessed mTOR-pathway activation by western-blot in a genetically unrelated control of similar age, the parents, the healthy brother and the patient (Fig. 3). However, we considered the healthy brother as the best control since he has not the variant and he is the closet genetically related person (47% coincidence in rare variants) to the patient in age (4 years older) [14]. As it has been published previously, there are two mTOR isoforms: the alpha-isoform corresponding to the full-length mTOR (mTORα, 2549 aa, ~260 kDa), and the shorter mTOR splicing beta-isoform (mTORβ, 706 aa, ~80 kDa) (Fig. 1d) [15]. The mTORβ has the ability to signal via phosphorylation of the characterized mTORα substrates (S6K1, AKT and 4EBP1), and it has also the potential to shorten the G1 phase of the cell cycle and to stimulate cell proliferation [15]. The mTORβ is the major isoform in blood (lymphocytes), being the levels of mTORα very low or even undetectable (Fig. 3a). Thus, the mTORα and p-mTORα were detected only in in the lymphocytes of control and mother (Fig. 3a). The study of the phosphorylation state of different members of the mTOR-pathway showed that the pathway was activated, as evidenced by the higher levels of p-mTORβ (Fig. 3b), p-4EBP1 (Fig. 3c), p-S6K1 (Fig. 3d, e), and p-AKT (Fig. 3f) in patient versus healthy brother. This activation was also evident respect to the age-matching control and parents, except in the case of p-4EBP1, where the levels in control and mother were higher (Fig. 3c). This higher level of p-4EBP1 in control and mother could be due to the presence of p-mTORα. Thus, p-mTORα could further increase the levels of phosphorylated downstream substrates (p-4EBP1, p-S6K1 and p-AKT) produced by p-mTORβ. In addition, p-mTORα could phosphorylate to 4EBP1 more efficiently that to the other two substrates. The phosphorylation state of other mTOR substrate, p-ULK1, was not detected in lymphocytes, although it could be detected in AD293 cells (Fig. 3a). A relationship between mTOR signalling and mitochondrial function has been described in mammalian cells, and this cross-talk is mediated by mtEF4 protein (mitochondrial translation elongation factor 4 or mitochondrial translation factor GUF1 –Q8N442-) [16]. Thus, the level of mtEF4 protein was reduced in the patient respect to her healthy brother (Fig. 4b), indicating that the translation of mtDNA-encoded RNA transcripts could be also reduced. With these data in hand, and taking in consideration that the patient has a combined complex I, III and IV deficiency (Table 1), several nuclear-encoded proteins (NDUFB8, SDHA, SDHB, UQCRC2, COX5A and ATP5A) and mtDNA-encoded proteins (MT-CO1 and MT-CO2) involved in oxidative phosphorylation (OxPhos) were measured (Fig. 4). There were not significant differences between the patient and her brother in the levels of NDUFB8 (complex I) (Fig. 4c), UQCRC2 (complex III) (Fig. 4f) and ATP5A (complex V) (Fig. 4j). However, a significant increase in SDHA (complex II) (Fig. 4d), SDHB (complex II) (Fig. 4e), COX5A (complex IV) (Fig. 4g), together with a decrease of MT-CO1 (complex IV) (Fig. 4h) and MT-CO2 (complex IV) (Fig. 4i) in the patient were observed.

Substrates phosphorylation analysis of mTOR-pathway by western-blot. a Lymphocytes from age-matching control (C), parents (F: father, M: mother), healthy brother (B) and patient (P) were lysed, separated by a gradient 4–20% (w/v) SDS-PAGE, transferred to PVDF membrane (Bio-Rad) and then probed with antibodies against the different proteins. The panels show a representative immunoblotting of mTOR, p-mTORS2448 or S605, p-4EBP1T37/46, p-S6KS371, p-S6KT389, p-ULK1S758, p-AKTS473 and β-actin. Densitometry analysis of the peak density normalized over β-actin of p-mTORβS605/mTORβ (b), p-4EBP1T37/46 (c), p-S6KS371 (d), p-S6KT389 (e), and p-AKTS473. Data were the mean (±SD) of three independent experiments, *P < 0.05 vs patient (P)

Analysis of mitochondrial protein by western-blot. a Lymphocytes from healthy brother and patient were lysed, separated by a gradient 4–20% (w/v) SDS-PAGE, transferred to PVDF membrane (Bio-Rad) and then probed with antibodies against the different proteins. The panels show a representative immunoblotting of mtEF4, NDUFB8 (nuclear-encoded complex I subunit), SDHA and SDHB (nuclear-encoded complex II subunits), UQCRC2 (nuclear-encoded complex III subunit), COX5A (nuclear-encoded complex IV subunit), MT-CO1 and MT-CO2 (mtDNA-encoded complex IV subunits), ATP5A (nuclear-encoded complex V subunit) and β-actin. Densitometry analysis of the peak density normalized over β-actin of mtEF4 (b), NDUFB8 (c), SDHA (d), SDHB (e), UQCRC2 (f), COX5A (g), MT-CO1 (h), MT-CO2 (i) and ATP5A (j). Data were the mean (±SD) of three independent experiments, *P < 0.05 vs Brother
Discussion
Herein, we describe a patient with macrocephaly, dysmorphic facial features, developmental delay, hypotonia, epilepsy, APS type I and defects on MRC activities (Table 1), suggesting a mitochondrial disease. However, molecular analysis of patient’s mtDNA ruled out depletion, rearrangements or known or putative affects function variants. Using whole exome sequencing (WES), we were able to identify a heterozygous variant in the nuclear-encoded gene MTOR. This missense variant (p.(Asp2412Val)) was found to be de novo (Fig. 1b) and new, since it has not been previously reported neither described in any genomic database (1000G, EVS, ExAC Browser and gnomAD browser). This novel variant is predicted to be “probably affects function” by in silico analysis software (SIFT, PolyPhen2, MutPrep and Mutation Taster). The amino acid is conserved among species (Fig. 1c) and it is located inside of Kinase Domain (KD) of the protein (Fig. 1d). The mTOR exists in two complexes, mTORC1 and mTORC2 [3], which regulate many metabolic processes, including protein synthesis, autophagy, lipid synthesis, energy metabolism, cell size, and others [4]. The mTORC1 signals to 4EBP1 and S6K that are key regulatory components of protein synthesis, by phosphorylation. Other functions of this complex are inhibition of autophagy (by phosphorylation of ULK1) and promoting ribosome biogenesis. By contrast, the mTORC2 is responsible for the phosphorylation of AKT and others kinases that regulate cytoskeletal organization. The functional consequences of p.(Asp2412Val) variant in mTOR was evaluated by substrates phosphorylation experiments. This study demonstrated that the change of the conserved aspartic acid in position 2412 to valine is a gain-of-function variant (GOF) since it was able to activate the mTOR pathway (Fig. 3) [17]. Thus, the phosphorylation of the mTORC1 substrates 4EBP1 and S6K, and phosphorylation of AKT, the substrate of mTORC2, were increased in patient, indicating activation of both complexes. Furthermore, the level of phosphorylated mTOR was higher in patient that in his healthy brother.
Activation of mTOR pathway drives energy consumption, principally by the protein synthesis machinery that is one of the most energy consuming processes in the cell [18]. But at the same time, it also positively regulates mitochondrial energy production by stimulating synthesis of nuclear-encoded mitochondrial proteins, including the components of the MRC complexes [19]. It has been hypothesized that mitochondrial respiratory capacity is regulated by the AKT/mTOR/4EBP1 cascade. Thus, the activation of AKT and/or inhibition of 4EBP1, both by phosphorylation, up-regulates the protein expression of all MRC complexes and increases OxPhos [20]. However, correct OxPhos function requires the coordinated expression of numerous genes encoded in two physically separated genetic systems, the nuclear (nDNA) and mitochondrial DNA (mtDNA) [21]. The crosstalk between mitochondrial and cytoplasmic translation is mainly regulated by mTOR pathway [19], being a major player in the regulation of mitochondrial translation the mtEF4 factor [16, 22]. The combined OxPhos deficiency in the MRC activity of the complexes I, III and IV in the patient muscle could be due to the loss of coordination between mitochondrial and cytoplasmic translation by the mTOR variant [4]. In consequence, the results using lymphocytes could explain the deficiency of complex IV (Fig. 4). Thus, while the nuclear-encoded mitochondrial proteins expression (i.e., SDHA and SDHB as components of complex II, and COX5A as component of complex IV) is increased as a consequence of mTOR pathway activation (Fig. 4d, e, g), the mitochondrial translation (as indicated by the lower level of mtEF4 (Fig. 4b)) and therefore the mtDNA-encoded proteins expression (i.e., MT-CO1 and MT-CO2 as a components of complex IV (Fig. 4h, i)) are reduced. However, the deficiencies of complexes I and III could not be justified based on the levels of the subunits measured in lymphocytes. Accordingly, the mTOR variant, in addition to its activation, must produce some change in the protein that could affect the mitochondrial translation by an unknown mechanism.
Deregulation of the mTOR pathway has been implicated in a diverse range of human pathologies (mTORopathies), including neurodegenerative and metabolic disorders [4]. Some variants in mTOR protein have been reported in patients presenting Smith-Kingsmore Syndrome (SKS), a multisystemic disorder characterized by ID, DD, megalencephaly and seizures that belongs to the group of mTORopathies. To date, only 10 variants in MTOR gene have been described in 27 families with SKS, and all of them are missense variant and suspected to result in GOF [6]. The symptoms of our patient are consistent with SKS, including the very common (ID, macrocephaly and seizures) and most of frequent findings (Table 2).
MRI findings in central nervous system had been found in most of individuals with SKS, including macrocephaly and hemi/megalencephaly, corpus callosum hypoplasia and ventricular dilatation [6]. In addition, our patient showed chronic ischemic lesions in cortical and sub-cortical areas suggestive of infarcts of small vessels (Fig. 2) [23]. This observation together with the hematological findings confirming the presence of anti-phospholipid antibodies (aPL), allowed the diagnosis of antiphospholipid syndrome type I (APS) in our patient [8]. It is interesting to note that the recruitment of the mTOR pathway has been described in many type of endothelium injuries including antiphospholipid antibodies [24]. The clinical picture (SKS) is in agreement with the genetic findings, but in addition, this is the first patient diagnosed with mTOR variant and APS.
Except for megalencephaly, the cerebral lesions in our patient are not imputable to cell grown and proliferation signals. The vascular origin of her lesions, as demonstrated by presence of hemosiderin in MRI sequences, is different respect to the principal lesions typical of other mTORopathies (tuberomas and white matter lesions). To further avoid brain damage, considering the evidences of mTOR pathway implication in the vascular lesions associated with APS, it was decided to start compassionate use of sirolimus, an mTOR inhibitor [9].
In summary, the mTOR variant produces a GOF of the protein, an activation of mTOR pathway that induces cell growth and proliferation responsible of SKS [5, 6, 25]. At the same time, the mTOR variant generates a loss of coordination between cytoplasmic protein synthesis and mitochondrial activity [19]. This leads to a combined OxPhos deficiency that could produces oxidative stress and apoptosis. The alteration of mitochondrial homeostasis by oxidative stress has been implicated in the pathophysiologic process that contributes to the development of aPL [26, 27]. Thereby, the oxidative stress induced the production of aCL and anti-β2GPI autoantibodies in lupus-prone mice (MRL/lpr) and in livers of TAL-deficient mice [26]. Furthermore, defects in OxPhos usually result in an accelerated apoptotic death of damaged cells [28, 29]. In turn, the presence of the mitochondrial-specific phospholipid cardiolipin (CL) on the surface of apoptotic cells could represent a trigger for antiphospholipid antibodies and activation of APS [30]. Thus, it has been proposed that while the presence of aPL is necessary but not sufficient to provoke a thrombotic event, an additional trigger (second hit hypothesis) is needed to initiate a vascular event in aPL carriers [31]. In our case, as it has been indicated above, the presence of CL on the surface of apoptotic cells could be this second hit.
Herein, we show data from only one patient, so further studies are needed to assess the relationship between mTOR variants and APS, and to demonstrate if it is a general mechanism in mTORopathies or our patient is a particular case. For this purpose, APS assessment (clinical, neuroradiologic and hematologic) should be investigated in patients with mTOR variants, in order to clarify the clinical relationship between mTOR, APS and endothelial injury [23].
In conclusion, our findings expand both the genetic and phenotypic spectrum of MTOR-associated diseases.
References
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–8.
Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43.
Russell RC, Fang C, Guan KL. An emerging role for TOR signaling in mammalian tissue and stem cell physiology. Development. 2011;138:3343–56.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Smith LD, Saunders CJ, Dinwiddie DL, Atherton AM, Miller NA, Soden SE, et al. Exome sequencing reveals de novo germline mutation of the mammalian target of rapamycin (MTOR) in a patient with megalencephaly and intractable seizures. J Genomes Exomes. 2013;2:63–72.
Gordo G, Tenorio J, Arias P, Santos-Simarro F, Garcia-Minaur S, Moreno JC, et al. mTOR mutations in Smith-Kingsmore syndrome: four additional patients and a review. Clin Genet. 2018;93:762–75.
Hughes GR. The antiphospholipid syndrome: 10 years on. Lancet. 1993;342:341–4.
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295–306.
Canaud G, Bienaime F, Tabarin F, Bataillon G, Seilhean D, Noel LH, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371:303–12.
Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Bertolaccini ML, Khamashta MA, Rodriguez-Ariza A, et al. Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood. 2012;119:5859–70.
Martinez B, del Hoyo P, Martin MA, Arenas J, Perez-Castillo A, Santos A. Thyroid hormone regulates oxidative phosphorylation in the cerebral cortex and striatum of neonatal rats. J Neurochem. 2001;78:1054–63.
Delmiro A, Rivera H, Garcia-Silva MT, Garcia-Consuegra I, Martin-Hernandez E, Quijada-Fraile P, et al. Whole-exome sequencing identifies a variant of the mitochondrial MT-ND1 gene associated with epileptic encephalopathy: west syndrome evolving to Lennox-Gastaut syndrome. Hum Mutat. 2013;34:1623–7.
Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra110.
Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–66.
Panasyuk G, Nemazanyy I, Zhyvoloup A, Filonenko V, Davies D, Robson M, et al. mTORbeta splicing isoform promotes cell proliferation and tumorigenesis. J Biol Chem. 2009;284:30807–14.
Cao X, Qin Y. Mitochondrial translation factors reflect coordination between organelles and cytoplasmic translation via mTOR signaling: Implication in disease. Free Radic Biol Med. 2016;100:231–7.
Hanai S, Sukigara S, Dai H, Owa T, Horike SI, Otsuki T, et al. Pathologic active mTOR mutation in brain malformation with intractable epilepsy leads to cell-autonomous migration delay. Am J Pathol. 2017;187:1177–85.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40.
Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. 2015;14:473–80.
Goo CK, Lim HY, Ho QS, Too HP, Clement MV, Wong KP. PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS ONE. 2012;7:e45806.
Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–68.
Gao Y, Bai X, Zhang D, Han C, Yuan J, Liu W, et al. Mammalian elongation factor 4 regulates mitochondrial translation essential for spermatogenesis. Nat Struct Mol Biol. 2016;23:441–9.
Zhu DS, Fu J, Zhang Y, Li SX, Zhang GX, Guan YT, et al. Neurological antiphospholipid syndrome: clinical, neuroimaging, and pathological characteristics. J Neurol Sci. 2014;346:138–44.
Gallo R, Padurean A, Jayaraman T, Marx S, Roque M, Adelman S, et al. Inhibition of intimal thickening after balloon angioplasty in porcine coronary arteries by targeting regulators of the cell cycle. Circulation. 1999;99:2164–70.
Negishi Y, Miya F, Hattori A, Johmura Y, Nakagawa M, Ando N, et al. A combination of genetic and biochemical analyses for the diagnosis of PI3K-AKT-mTOR pathway-associated megalencephaly. BMC Med Genet. 2017;18:4.
Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheuma. 2016;68:2728–39.
Ioannou Y, Zhang JY, Qi M, Gao L, Qi JC, Yu DM, et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen beta2-glycoprotein I. Arthritis Rheum. 2011;63:2774–82.
Song HP, Chu ZG, Zhang DX, Dang YM, Zhang Q. PI3K-AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by MitoKATP-mediated mitochondrial translocation of pAKT. Cell Physiol Biochem. 2018;49:717–27.
Schull S, Gunther SD, Brodesser S, Seeger JM, Tosetti B, Wiegmann K, et al. Cytochrome c oxidase deficiency accelerates mitochondrial apoptosis by activating ceramide synthase 6. Cell Death Dis. 2015;6:e1691.
Manganelli V, Capozzi A, Recalchi S, Signore M, Mattei V, Garofalo T, et al. Altered traffic of cardiolipin during apoptosis: exposure on the cell surface as a trigger for “antiphospholipid antibodies”. J Immunol Res. 2015;2015:847985.
Ceccarelli F, Chighizola C, Finazzi G, Meroni PL, Valesini G. Thromboprophylaxis in carriers of antiphospholipid antibodies (APL) without previous thrombosis: “Pros” and “Cons”. Autoimmun Rev. 2012;11:568–71.
Acknowledgements
The authors thank the patient and her family for their contribution, and Alicia Torrado for valuable comments on the manuscript. This work was supported by the Spanish Instituto de Salud Carlos III (ISCIII) and European Regional Development Fund (ERDF) (grant PI14/00790 and PI17/00487 to FMA). FJCV was supported by fellowship from the Instituto de Investigación Hospital 12 de Octubre (i+12). PCR and LHS were supported by fellowships from Comunidad de Madrid and European Social Fund (ESF) (PEJ16/MED/TL-1655 and PEJD-2017-PRE-BMD-4883 respectively). MERG was supported by fellowship from ISCIII and ERDF (grant PI14/00790 and PI17/00487).
Author information
Authors and Affiliations
Contributions
All authors contributed significantly to this research. MB, AMA, and EMH clinically characterized the patient. MERG, FJCV, LHS and PCR performed the experiments. FMA planned and supervised the study, wrote the manuscript and provided the funding.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rodríguez-García, M., Cotrina-Vinagre, F., Bellusci, M. et al. A novel de novo MTOR gain-of-function variant in a patient with Smith-Kingsmore syndrome and Antiphospholipid syndrome. Eur J Hum Genet 27, 1369–1378 (2019). https://doi.org/10.1038/s41431-019-0418-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0418-1
This article is cited by
-
An immunogenomic exome landscape of triple positive primary antiphospholipid patients
Genes & Immunity (2024)
-
Smith-Kingsmore syndrome with nystagmus as the initial symptom
Acta Epileptologica (2023)
-
Genetics of Antiphospholipid Syndrome
Current Rheumatology Reports (2019)
