
Abstract
With exome/genome sequencing (ES/GS) integrated into the practice of medicine, there is some potential for reporting incidental/secondary findings (IFs/SFs). The issue of IFs/SFs has been studied extensively over the last 4 years. In order to evaluate their implications in care organisation, we retrospectively evaluated, in a cohort of 700 consecutive probands, the frequency and burden of introducing the search for variants in a maximum list of 244 medically actionable genes (genes that predispose carriers to a preventable or treatable disease in childhood/adulthood and genes for genetic counselling issues). We also focused on the 59 PharmGKB class IA/IB pharmacogenetic variants. We also compared the results in different gene lists. We identified variants (likely) affecting protein function in genes for care in 26 cases (3.7%) and heterozygous variants in genes for genetic counselling in 29 cases (3.8%). Mean time for the 700 patients was about 6.3 min/patient for medically actionable genes and 1.3 min/patient for genes for genetic counselling, and a mean time of 37 min/patients for the reinterpreted variants. These results would lead to all 700 pre-test counselling sessions being longer, to 55 post-test genetic consultations and to 27 secondary specialised medical evaluations. ES also detected 42/59 pharmacogenetic variants or combinations of variants in the majority of cases. An extremely low metabolizer status in genes relevant for neurodevelopmental disorders (CYP2C9 and CYP2C19) was found in 57/700 cases. This study provides information regarding the need to anticipate the implementation of genomic medicine, notably the work overload at various steps of the process.
Similar content being viewed by others

Introduction
The National Human Genome Research Institute defines genomic medicine as “an emerging medical discipline that involves using genomic information about an individual as part of their clinical care (e.g., for diagnostic or therapeutic decision-making) and the health outcomes and policy implications of that clinical use.” Given its potential to revolutionise the practice of medicine, with an expected economic impact within the next decade, genomic medicine is receiving international attention and significant media coverage. The field has been boosted in the last decade by the development of ground-breaking sequencing technologies commonly referred to as next-generation sequencing (NGS).
The recent implementation of exome sequencing (ES) for the diagnosis of rare and heterogeneous Mendelian disorders showed a diagnostic yield of > 25%, making it one of the most powerful individual diagnostic test for such conditions with intellectual and/or developmental abnormalities [1,2,3,4,5,6,7]. Smaller studies suggest that genome sequencing (GS) may increase the diagnostic yield to 60% in individuals with developmental anomalies and intellectual disability (ID) [8,9,10,11]. Implementing ES and GS in the clinical setting is a great challenge. With the increasing application of clinical ES and GS, it is necessary for geneticists and other healthcare providers to understand the benefits and limitations in order to interpret the clinical relevance of genomic variants identified in the context of health and disease. Collaborations with specialists across diverse disciplines and patients will undoubtedly be key attributes of the future practice of clinical genetics.
Beyond the identification of variants affecting protein function for the diagnosis of Mendelian disorders, the sequencing of individual genomes can detect numerous variants that may be relevant for clinical care and thus enable clinical interventions to improve future health outcomes in patients and their at-risk relatives, from predicting late-onset genetic disorders accessible to prevention or treatment, to implications for genetic counselling and differential drug responses and toxicities. These data can be particularly relevant in patients with rare disorders that may require multiple medications. Such variants were first referred to as incidental findings (IF) in the North-American literature [12], but the term secondary findings (SF) is now recommended in the literature because the term IF was not appropriate if the variants were being actively searched for [13,14,15,16]. The American College of Medical Genetics and Genomics (ACMG) initially developed a minimum list of 56 genes considered medically actionable to be reported by all clinical laboratories performing NGS analyses [12], recently revised to 59 genes [15], unless patients opt out (American College of Medical Genetics and Genomics 2014). “Actionable” genes are defined as having deleterious mutation(s), whose penetrance would result in specific, defined medical recommendation(s), both supported by evidence and, when implemented, expected to improve an outcome(s) in terms of mortality or the avoidance of significant morbidity [12,13,14,15,16,17]. Some clinical laboratories have returned a smaller or a broader set of IFs/SFs [17,18,19,20], and some publications have proposed methods for evaluating the clinical actionability of IFs/SFs from genome-scale sequencing [21, 22]. Moreover, some pharmacogenetic alleles are recognised to induce toxicity or adverse effects or reduce the efficacy of drugs, and guidelines are being developed to modify drug prescription with regard to pharmacogenetic polymorphisms [23,24,25]. The position of the European society of Human Genetics (ESHG) is quite different from the position of the ACMG, since the ESHG has preferred to recommend the use of panels (bioinformatic or not) to the prescription of ES to avoid the question of IFs/SFs [16]. Indeed, the question of psychological burden of such information, as well as the limited information on clinical utility and reduced penetrance should not be neglected.
Analysing all these variants, if the patients or caregivers agree, can be a step towards more general implementation of genomic medicine. However, interpreting such variants beyond the primary cause of the disease can be challenging and time-consuming for both clinicians and labs. Indeed, it requires additional clinical (pre-test counselling information, post-test counselling in positive cases, information to relatives and specialised consultations in some cases) and lab work. This study aimed to evaluate the frequency of actionable IFs/SFs from a specific longer list that included a variety of different types of genes and variants accessible to treatment or prevention (late-onset diseases, genetic counselling, pharmacogenetics) in a series of 700 exomes, in order to evaluate the impact on the organisation of care.
Materials and methods
Development of the list of genes and pharmacogenetic polymorphisms
The “disease” gene list, referred to as the “maximum list”, was developed to gather actionable genes from various previously published lists of genes [12, 15, 17,18,19,20], with a significant impact for the patient and/or family in terms of prevention, treatment or genetic counselling (Fig. 1a). A list of 244 genes of interest was defined by an expert panel (Table 1) in two categories: (1) genes that predispose to a disease in childhood or adulthood accessible to prevention or treatment. It includes 59 genes recommended for return by the American College of Medical Genetics and Genomics (ACMG) guidelines, 69 additional actionable genes associated with medically actionable genetic conditions that might remain undiagnosed in adults as defined by Amendola [18] and 4 additional genes predisposing to coagulation disorders not included in the previous lists (F2, F8, F9, VWF); this list comprised 100 genes with an autosomal dominant, 2 with an autosomal semi-dominant, 24 with an autosomal recessive and 6 with an X-linked mode of inheritance. (2) Genes responsible for autosomal or X-linked recessive disorders, for which information about the heterozygote or carrier status could be important for genetic counselling. This list comprises three genes responsible for a recessive disease with a high frequency of heterozygotes in France, thus justifying the genetic screening of relatives (CFTR, SMN1, CYP21A2). Indeed, when a variant affecting or probably affecting protein function has been detected in one member of a couple in one of these genes, an access to partner screening will be offered and covered by French health insurance. This list also comprises 110 genes implicated in X-linked intellectual deficiency (XLID) [26]. In the majority of genes, all variants were considered, whereas in a few genes, only specific known causal variants were considered (32 CFTR variants of the Elucigene® kit, F2-rs1799963:G>A (hg19 chr11:g.46761055G>A NG_008953.1:g.25313G>A—NM_000506.4:c.*97G>A), F5-rs6025:C>T (hg19 chr1:g.16951949G>A, NG_011806.1:g.41721G>A—NM_000130.4:c.1601G>A) (FV Leiden variant/only homozygous status) and SMN1 deletion).

The results were also analysed in parallel using the other most commonly referenced list, including the ACMG list, “Dorschner’s list” [17], “Amendola’s list” [18], “Dewey’s list” [19] and the “100,000 genomes’ list” [20], in order to have a comparison of frequencies in the exact same cohort of patients.
The list of pharmacogenetic polymorphisms was based on variant–drug associations listed in the Pharmacogenomics Knowledgebase (PharmGKB). To delineate the impact of identifying relevant pharmacogenetic IFs/SFs, we collected 59 variants or combinations of variants from the PharmGKB 1A/1B categories because of the substantive evidence for clinical relevance (Table 2).
Bioinformatics analysis from ES analysis
We specifically looked for genomic variants in the determined gene list among the results from ES performed in 700 consecutive probands, mainly Caucasian, for research or diagnosis in a context of developmental abnormalities. We used our usual procedures for ES, bioinformatics analysis and variant annotation [7]. However, the pipeline had to be adapted to remove the filter that excluded non-rare variants (c.f. online-only material). Some variants were known to be undetectable, such as F2-rs1799963:G>A (hg19 chr11:g.46761055G>A NG_008953.1:g.25313G>A—NM_000506.4:c.*97G>A) located in the non-exonic 3′ UTR region or SMN1 deletion because of SMN2 paralog gene. Most CYP21A1 variants responsible for the classical form of congenital adrenal hyperplasia were also undetectable because of gene conversion. Only 61% of these high-priority pharmacogenetic variants were adequately covered by ES for reporting (PharmGKB 1A/1B categories), due to undetectable intronic variants and CNVs, pseudogenes or small CNVs. Besides the number of patients with at least one pharmacogenetic variant, we especially focused on alleles or combinations of alleles leading to an extreme phenotype (poor metaboliser) in genes of interest in drugs usually prescribed in patients with neurodevelopmental disorders (CYP2C9 and CYP2C19).
Information regarding variant calling can be found in the Supplementary Material. No Sanger confirmation was done.
Criteria for variant annotation and classification
We aimed to classify each variant as a variant affecting protein function, a variant probably affecting protein function, a variant of uncertain significance (VUS), a likely benign variant or a benign variant. We initially looked for all variants with a known Clinvar status, and considered pathogenic, likely pathogenic and discordant variants. When the ClinVar classification was discordant, we based our criteria on the evidence outlined in the recommendations of the ACMG and Association for Molecular Pathology (ACMG/AMP), available in the literature and databases [27]. The criteria are detailed in the online-only material. Like Amendola, we tried to evaluate the time spent by the biologists on the interpretation, literature review and categorisation step for each variant. Indeed, the first biologist systematically interpreted every variant. When interpretation required discussion, a second biologist was implicated in the interpretation and/or the reference diagnosis labs were contacted, if necessary. Time for variant interpretation was systematically recorded.
Only variants affecting or probably affecting protein function were returned to patients. However, some variants were not considered for return to patients in the absence of a combination that led to a significant risk of disease. These included CYP21A2 variants responsible for a non-classic mild type of congenital adrenal hyperplasia in 43 individuals with the predominant well-known CYP21A2-rs6471 variant (hg19 chr6:32007887G>C, NG_007941.3:g.6806G>C—NM_000500.7:c.844G>C—p.(V282L)) (30/31 cases), F5-rs6015:C>T in the absence of a homozygous status or a combination with other predisposition factors, homozygous for the PI allele (hg19 chr14:g.94847262A>T, rs17580:A>T) leading to no clinical consequences, and CFTR variants responsible for a non-classic type of CF, including the CFTR-rs78655421 variant (hg19 chr7:g.117171029G>A—NG_016465.4:g.70192G>A— NM_000492.3:c.350G>A—p.(Arg117His)) because no genetic counselling is recommended for this variant in the French or usual recommendations [28].
For each pharmacogenetics polymorphism or combination of polymorphisms, we used the well-known annotation from the PharmGKB 1A/1B categories: 1A for a variant–drug combination in a Clinical Pharmacogenetics Implementation Consortium (CPIC) or medical society-endorsed PGx guideline; 1B for a variant–drug combination because the preponderance of evidence shows an association. PGx information was not available for some genes (CYP2D6, CYP3A5, VKORC1), because ES does not detect non-exonic or CNV alleles. Indeed, to be relevant, the PGx information for one gene needs the determination of different variants, also implicating the detection of multiallelic variants to determine the phenotype: the ultra-rapid metabolizer, extensive metabolizer, normal or poor metabolizer phenotype. We determined the total proportion of patients carrying PGx actionable variants or combinations of variants that could be determined by ES in order to identify the limits of WES for such predictions (Table 2), and also the percentage of individuals with a poor metabolizer (PM) status in the CYP2C9 and CYP2C19 genes, which could lead to adverse effects or the adjustment of drug dosages potentially prescribed in patients with neurodevelopmental disorders (anti-epileptics, antidepressants and psychotropic medications, Table 2). This included patients with homozygous or compound heterozygous rs1799853:C>T (hg19 chr10:96702047C>T) CYP2C9*2 and/or rs1057910:A>C (hg19 chr10:g.96741053A>C) CYP2C9*3, and patients with homozygous or compound heterozygous rs4244285:G>A (chr10:96541616G>A) CYP2C19*2, rs4986893:G>A (hg19 chr10:g.96540410G>A) CYP2C19*3 and/or rs28399504 CYP2C19*4:A>G (hg19 chr10:g.96522463A>G). CYP2D6 phenotype was not determined because this classification requires the analysis of multiallelic combination and, in fact, some genetic polymorphisms (CYP2D6*5,>2N) were actually not detected by ES.
Medical recommendations
To evaluate the impact of introducing the search for IFs/SFs on the care of patients as a more generalised implementation of genomic medicine, the repercussions on patient care were listed (c.f. online-only material) and compared with previously published lists of actionable genes [12, 15, 18,19,20]. For pharmacological variants, indications and drug names associated with the impact of each variant were taken from the PharmGKB dataset (Table 2).
Results
Bioinformatics results
The 700 consecutive probands comprised 371 males and 329 females, with a large majority of children and Caucasians presenting with ID or multiple congenital anomalies. No particular bioinformatics problems were found for the genes belonging to the category 1 list. However, difficulties were encountered in detecting heterozygotes of some recessive disorders, including, for example, CFTR-rs113993960 variant (hg19 chr7:g.117199646-117199648delCTT—NG_016465.4:g.98809_98811delCTT—NM_000492.3:c.1521_1523delCTT—p.(Phe508del)), which required adjustments of the bioinformatics because of misalignment. For the F5-rs6025:G>A variant (hg19 chr1:g.169519049G>A—NG_011806.1:g.41721G>A—NM_000130.4:c.1601G>A), the wild-type allele is considered the mutated allele in the reference genome, which also required adjustments of the bioinformatics because of abnormal annotation.
Variants interpretation and classification
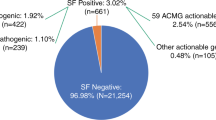
Within the “maximum list” of genes, the Clinvar database identified 20 pathogenic or likely pathogenic variants and 42 discordant variants in addition to 11 truncating variants with no Clinvar status, in 45 different genes, in 148/700 index cases (Supplementary Table 1). Following the interpretation recommendations of the ACMG/AMP, we reclassified these 73 variants as 28/73 variants affecting or probably affecting protein function, 31/73 VUS and 14/73 benign/likely benign variants (Table 3; Fig. 2; Supplementary Table 2). Twenty-eight variants affecting or probably affecting protein function were present in 55/700 index cases (7.8%).

Scheme of interpretation results of the 73 variants among the 244 genes and variants of interest, from the cohort of 700 cases, and their repercussions on patient care
For each variant, the process of fine interpretation required from <5 min to 30 min, depending on the data available in public databases and the medical literature. For example, for the 32 CFTR variants of the Elucigene® kit, as well as the F5-rs6025: G>A variant (hg19 chr1:g.169519049G>A—NG_011806.1:g.41721G>A—NM_000130.4:c.1601G>A) and HFE-rs1800562:G>A (hg19 chr6:g.26093141G>A—NG_008720.2:g.10633G>A—NM_000410.3:c.845G>A), with a well-known pathogenic status, interpretation took <5 min and did not require a second biologist. For other variants with Clinvar pathogenic status and previously published as causal in similar phenotypes, the time needed for variant interpretation, Sanger chromatogram verification and additional specific biological report was usually around 15 min in order to check specific databases and/or literature data to check the status accuracy. Variants with discordant Clinvar status and frameshift variants not reported in Clinvar induced more investigations in the literature taking on average ~30 min for the first biologist and an additional discussion of 15 min with the second biologist. Mean time for the 700 patients was about 6.3 min per patient for genes that predispose to a disease in childhood or adulthood accessible to prevention or treatment, and 1.3 min per patient for genes for genetic counselling, with a mean time of 37 min for the variants that had to be reinterpreted or subjected to Sanger chromatogram verification and an additional specific biological report. Contacting a referent lab when necessary was planned in the study design, but was not needed in any case.
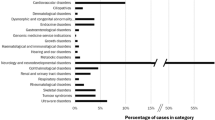
The distribution of variants that could be returned if requested by the patients was as follows: In the first category of genes able to cause a disease that may be accessible to treatment or prevention later in life, we identified 26 cases (3.7%) harbouring 21 different variants affecting or probably affecting protein function (14 cases (2%) and 13 cases (1.9%) with variants in the 56 and 59 ACMG actionable genes, respectively [12, 15], with a smaller percentage in ACMG59, explained by the removal of the MYLK gene, although four new genes were included in this gene list. Among these 26 cases, we found 25 cases with an autosomal dominant heterozygous variant (3.6% cases) and one case with autosomal recessive biallelic variants (HFE) (0.1% cases). Among the 700 probands, autosomal dominant diseases included hereditary cardiac disease (11 cases, 1.6% cases), cancer predisposition (6 cases, 0.9% cases), Von Willebrand disease (6 cases, 0.9% cases), acute intermittent porphyria (1 case, 0.1% cases) and Dopa-responsive dystonia (1 case, 0.1% cases) (Fig. 2). When referring to the other gene lists, we found 20 cases in Dorschner’s list (2.9%), 18 in Amendola’s list (2.6%), 16 cases in Dewey’s list (2.3%) and 4 cases in the 100,000 genomes project list (0.6%) [18,19,20]) (Fig. 1).
For the second category of genes for which heterozygote status can be of importance in terms of genetic counselling, we identified variants to be returned in 29 individuals (4.1%), including 26 individuals with CTFR variants (22/26 individuals with the predominant CFTR-rs113993960 variant (hg19 chr7:g.117199646-117199648delCTT—NG_016465.4:g.98809_98811delCTT—NM_000492.3:c.1521_1523delCTT—p.(Phe508del)) and 4/21 individuals each with a different CFTR heterozygous variant), one individual with a CYP21A2 variant that could be responsible for a classic type of congenital adrenal hyperplasia, one female ARX carrier and one female GPC3 carrier (Fig. 2).
Among the 700 probands, 695 presented with potential pharmacogenetic variants or combinations of variants despite only 39/59 detectable high-priority variants or combinations of variants (PharmGKB 1A/1B categories). To determine whether variants or combinations of variants were of potential therapeutic relevance in this population, we focused on the pharmacogenetic variants or combinations of variants that might provide insights into their response to a prescribed medication for neurodevelopmental disorders (behavioural disturbances, depression or seizures, Table 2). This led to the identification of five pharmacogenetically relevant variants or combinations of variants implicating the CYP2C9 and CYP2C19 cytochromes (no prediction possible for the CYP2D6 cytochrome, due to the multiallelic nature of the at-risk genotype). We only focused on extreme phenotypes that could lead to a prescription adjustment. Finally, 12 patients were homozygous for CYP2C9*2:C>T, 6 for CYP2C9*3:A>C and 17 were compound heterozygous for CYP2C9*2:C>T and CYP2C9*3A>C. These genotypes are associated with a “slow metabolizer” phenotype. Also, 21 patients were homozygous for CYP2C19*2:G>A, and 1 compound heterozygous for CYP2C19*2:G>A and CYP2C19*3:G>A. In total, 57 patients could be considered as at risk of adverse effect when they received drugs metabolised mainly by hepatic cythochromes (CYP2C9 or CYP2C19) (8.1%).
Potential downstream results of secondary variant reporting
If returned to cases, SFs would extend the time needed for pre-test counselling (Fig. 2). For genes able to cause a disease that may be accessible to treatment or prevention later in life, SFs (3.9%) would have led to 26 post-test genetic consultations, as well as 11 cardiology, 6 haematology, 4 gynaecology, 1 dermatology, 1 gastroenterology, 1 oncology, 1 neurology and 1 metabolism screening, usually on a yearly basis. Annual imaging and biological investigations would also likely have been recommended for these patients. Also, some patients with reported SF, in particular in those with cardiovascular risk, would have access to medication and/or medical devices (Table 3). Additional family evaluations should also be anticipated, but are difficult to project.
For the second category of genes, the heterozygote status can be of importance in terms of genetic counselling. Here, genetic counselling would have been recommended in 29 cases, including CF, a classic type of congenital adrenal hyperplasia, ARX syndrome and Simpson–Golabi–Behmel syndrome, and would have led to further consultations for at-risk relatives and partners. The latter number of additional consultations is difficult to project and depends on the family composition.
Discussion
The issue of IFs/SFs has been studied extensively over the last 4 years. The difficulty in interpreting additional IFs/SFs has already been evaluated according to the ACMG 59 genes minimum list [15, 18, 29], but the impact on patient care and the organisation of healthcare has not been clearly evaluated. Concerns have been raised about the financial, structural and organisational challenges that may delay the introduction of genomic medicine [30].
For this purpose, we have chosen to systematically identify and interpret clinically relevant variants predicted to cause a genetic disease accessible to prevention and treatment in the individual’s lifetime, or a heterozygote status of frequent recessive disorders, accessible to heterozygote status screening in relatives, or carriers of X-linked recessive disorders for genetic counselling issues. The question of the list and criteria for the clinical actionability of genes has been studied by others, and in particular the Clinical Genome Resource (ClinGen), in an effort to pave the way for precision medicine. They have developed a practical, standardised protocol to identify available evidence of actionability for disorders and associated genes [22]. Regarding the list of actionable genes leading to predisposition to a later-onset disease, we have made the choice of using the ACMG list, but also to extend to other lists that have been published in order to evaluate the impact of the gene list on the results, in a situation where results are not given back to patients. However, the level of medical actionability and penetrance have not been made with the same definition and protocol, which requires attention before transferring to a diagnosis context. Regarding the list of genes for genetic counselling, our choice to restrict our research to three genes in which we searched for heterozygote status is potentially disputable, and one could argue that every recessive disease gene has some genetic counselling aspect. For example, other studies that looked at all recessive heterozygote status found a median of two variants affecting or probably affecting protein function per patient [31]. However, the screening of partners would be not accessible in the majority of cases in the French healthcare system. Our choice of genes takes into account the effective actionability for patients.
The first finding of this study is the information it provided in terms of feasibility. The detection of heterozygotes for some recessive disorders was difficult, making some bioinformatics tool adjustments necessary. SMN1 deletion and the majority of CYP21A1 variants responsible for the classical form of congenital hyperplasia could not be detected. It was also sometimes difficult to draw conclusions in diseases following an autosomal recessive mode of inheritance, because the bioinformatics analyses did not allow us to determine whether the variants were in cis or trans. Twenty variants or combination of variants from Pharm GKB 1A/1B categories could not be detected because ES cannot detect intronic and somatic variants, as well as some CNVs (Table 2).
The second finding is the frequency of variants found in each category, which is necessary to evaluate the need for genomic medicine in terms of the future organisation of healthcare. Interestingly, we determined the frequency of SFs in the same cohort, depending on the gene list that had been chosen. An actionable variant was identified in 0.6–3.7% of cases. These included 1.9% cases (13 variants) in the ACMG 59 genes list. Hereditary cardiac disease was the most frequent, but was not present in all of the gene lists, such as the 100,000 genomes project SFs gene list. The frequency of SFs for autosomal dominant genes with a predisposition for late-onset diseases was higher than those published in the data of 112 genes selected by an expert panel as associated with medically actionable genetic disorders in 4300 European- and 2203 African-ancestry adults with an undiagnosed disease sequenced by the NHLBI Exome Sequencing Project (ESP). Among European-ancestry participants, 2% had a SNV, affecting or probably affecting protein function, and the percentage was even lower in participants with African ancestry (1.2%) [18]. Within the same gene list, a variant affecting or probably affecting protein function was found in 2.7% of patients in our cohort (Fig. 1). The results for category 2 were mainly represented by heterozygote status for CFTR variants of the Elugene kit in 3.7% of the patients.
The impact of the additional economic burden for deploying the search for SFs/IFs on organisation of care should not be neglected. Our results suggest that there would be need for additional costs, including the extent of the time for pre-test information, post-test information, specialised clinical encounters and genetic counselling for at-risk relatives in positive cases. It should be stressed that, ideally, consultations to inform patients about the implications of SFs/IFs should be different from medical consultations, and led by a genetic counsellor in order to have enough information to obtain informed consent. Recommendations to guide the integration of genomics into clinical practice have been discussed, and they take into account the fact that enlarging the analysis of the exome will result in IFs/SFs that will change the scope of practice for clinical geneticists [32]. Amendola’s study estimated that the time spent by the initial biologist on the literature review and categorisation step for each HGMD disease-causing variant was up to 37 min. For double-reviewed variants, 53% of the classifications were discrepant, thus underlining the need for a better curated variant interpretation database [18]. The time our team spent on returning SFs could be estimated at 6.3 min per patient for genes that predispose to a disease in childhood or adulthood and are accessible to prevention or treatment, and 1.3 min per patient for genes for genetic counselling. However, a mean of 37 additional minutes per variant was required for the few variants that had to be reinterpreted. This result should be taken with caution since time spent could be a function of the level of expertise and interest of the biologist. Also, the fact that the study used anonymous data, implying that results were not given back to patients, could have led to an underestimation of the need for advice by a referent lab for adequate interpretation. The choice of using the ClinVar classification of variant as a first filter, and to reevaluate variants with interpretation differences can be disputable. A considerable effort is made in the international community to assess the medically significant differences in variant classification, that should improve this approach in the future, and improve the care of patients with, or at risk for, genetic disorders [33,34,35].
An interesting study used a quantitative framework to evaluate the economic consequences of returning ACMG-recommended IFs/SFs to individuals receiving genomic sequencing [36]. The authors concluded that returning IFs/SFs is cost-effective for certain patient populations, but not cost-effective in generally healthy individuals, unless NGS costs <$500. The assessment of pharmacogenetic variants or combinations of variants in this series confirmed the limit of ES for accurate pharmacogenetic counselling. Indeed, as we detected only 61% of the high-priority SNPs (Pharm GKB 1A/1B categories), and as multiallelic combinations are not rare, the interpretation of the risk when prescribing medication was sometimes difficult. In order to overcome this issue, the National Institutes of Health (NIH) Undiagnosed Diseases Program (UDP) used SNP chip analysis in addition to ES to evaluate the frequency and the therapeutic informativeness of pharmacogenetic IFs/SFs annotated in the Pharmacogenomics Knowledgebase (PharmGKB) sequence variants or combination of variants within a cohort of 1101 individuals. Interestingly, medication records of participants were used to identify individuals with prescribed medications even though they had a genetic variant that might alter efficacy. Nineteen PharmGKB 1A/1B variants or combinations of variants from the list used at the time of the work were identified in SNP chip sequence data, and 21 PharmGKB 1A/1B variants or combinations of variants were identified in the exome-sequence data using the TruSeq kit (Illumina). Only nine participants had pharmacogenetic IFs/SFs associated with altered efficacy of a prescribed medication, demonstrating that some pharmacogenetic IFs could be potentially useful for guiding therapy, and could be worth reporting [37]. However, another study that interviewed 159 Mayo Clinic clinicians revealed that half of them were uncomfortable with the inclusion of pharmacogenomic data in primary care. It highlights the importance of increasing the awareness of healthcare providers about such progress [38]. The frequency of the pharmGKB pharmacogenetic alleles found in this study is not unexpected, since for example Parsons et al. found a median of one pharmGKB SNP per clinical exome, though using a much smaller list of SNPs [31].
In our study, we chose to focus particularly on pharmacogenetic variants or combinations of variants relevant for neurodevelopmental disorders (e.g., behavioural disturbances, depression or seizures) [39, 40], because the indication for ES in the 700 patients were mainly developmental disorders with frequent neurodevelopmental phenotypes. These explorations implicate the analysis of the CYP2C19, CYP2C9 and CYP2D6 variants or combinations of variants. Unfortunately, our results were limited by the non-determination of all CYP2D6 variants. Another Mayo Clinic study outlined the necessity of using a custom genotyping method to determine all CYP2D6 variants or combinations of variants [37]. If we do not focus on the extreme alleles, 415/700 different patients had at least one listed variant or combination of variants from the PharmGKB 1A category within the CYP2C19 and CYP2C9 gene, relevant to toxicity or variability of pharmacokinetics or pharmacodynamics of different medications (Table 2). If we focus on extreme patients, 56 patients were found to be poor metabolizers for CYP2C9 and CYP2C19. These results indicate that patients could have a more appropriate treatment when the drug has to be prescribed and they could experience fewer adverse drugs-related events. The list of drugs for neurodevelopmental disorders most likely to be impacted by the SNPs analysed here can be found in Table 2 and includes anti-epileptic medications as well as antidepressants. The issue of prospectively interrogating pharmacogenetic variants regarding a specific medication rather than giving a list of pharmacogenetic variants or combinations of variants in the ES/GS report remains to be determined, especially since physicians have to learn how to manage pharmacogenetic findings [37]. Considering the limit of pharmacogenomic predictions from ES, the use of GS in the future should overcome this issue. Also, the dosing recommendations are currently only for adults for the vast majority of these SNPs, which may curb their utility in children with neurodevelopmental disorders.
Some limitations of the study could be argued. Our population of patients was selected and mainly Caucasian, so it is difficult to know if the results are broadly applicable to other populations. For example, the average time for variant classification would likely be much longer when analysing data from a more heterogeneous patient population because most variant databases are issued from limited populations. The question of the list of actionable genes to be studied in children could be debated since the general rules for genetic testing in minors do not authorise genetic testing at a paediatric age for adult-onset diseases that cannot be prevented in childhood. Finally, this study did not concentrate of the impact of these findings on patients, in a meaningful way or with adverse effects of the provision of this information.
In conclusion, this study highlights the need to reorganise healthcare if genomic medicine is implemented for developmental diseases, particularly the analysis of further information available in NGS data. However, the extent of the actionable gene list with regard to medicoeconomic and ethical issues warrants additional discussion.
URLs
PharmGKB, https://www.pharmgkb.org; The 100,000 Genomes Project Protocol v3, Genomics England; https://www.genomicsengland.co.uk/100000-genomes-project-protocol/
References
De Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11.
Rauch A. Exome sequencing in unspecific intellectual disability and rare disorders. Mol Cytogenet. 2014;7(Suppl 1 Proceedings of the International Conference on Human):I26.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704.
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
Nambot S, Thevenon J, Kuentz P, Duffourd Y, Tisserant E, Bruel AL, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med. 2018;20:645–54.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
Smith LD, Willig LK, Kingsmore SF. Whole-exome sequencing and whole-genome sequencing in critically Ill neonates suspected to have single-gene disorders. Cold Spring Harb Perspect Med. 2015;6:a023168.
Petrikin JE, Willig LK, Smith LD, Kingsmore SF. Rapid whole genome sequencing and precision neonatology. Semin Perinatol. 2015;39:623–31.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013;21:S1–5.
Allyse M, Michie M. Not-so-incidental findings: the ACMG recommendations on the reporting of incidental findings in clinical whole genome and whole exome sequencing. Trends Biotechnol. 2013;31:439–41.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for Diagnostic next-Generation Sequencing. Eur J Hum Genet. 2016;24:2–5.
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93:631–40.
Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305–15.
Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:6319.
The 100, 000 Genomes Project Protocolv3, Genomics England. Page 43, https://doi.org/10.6084/m9.figshare.4530893.v2. 2017. https://www.genomicsengland.co.uk/100000-genomes-project-protocol/
Berg JS, Foreman AK, O’Daniel JM, Booker JK, Boshe L, Carey T, et al. A semiquantitative metric for evaluating clinical actionability of incidental or secondary findings from genome-scale sequencing. Genet Med. 2016;18:467–75.
Hunter JE, Irving SA, Biesecker LG, Buchanan A, Jensen B, Lee K, et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med. 2016;18:1258–68.
Chang MT, McCarthy JJ, Shin J. Clinical application of pharmacogenetics: focusing on practical issues. Pharmacogenomics. 2015;16:1733–41.
Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharm Ther. 2013;93:324–25.
Lindor NM, Thibodeau SN, Burke W. Whole-genome sequencing in healthy people. Mayo Clin Proc. 2017;92:159–72.
Piton A, Redin C, Mandel JL. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet. 2013;93:368–83. Erratum in: Am J Hum Genet. 2013;93:406.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Thauvin-Robinet C, Munck A, Huet F, de Becdelièvre A, Jimenez C, Lalau G, et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J Med Genet. 2009;46:752–8.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016;98:1067–76.
Budin-Ljøsne I, Harris JR. Patient and interest organizations’ views on personalized medicine: a qualitative study. BMC Med Ethics. 2016;17:28.
Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2:616–24.
Bowdin S, Gilbert A, Bedoukian E, Carew C, Adam MP, Belmont J, et al. Recommendations for the integration of genomics into clinical practice. Genet Med. 2016;18:1075–84.
Bennette CS, Gallego CJ, Burke W, Jarvik GP, Veenstra DL. The cost-effectiveness of returning incidental findings from next-generation genomic sequencing. Genet Med. 2015;17:587–95.
Harrison SM, Dolinksy JS, Chen W, Collins CD, Das S, Deignan JL, et al. Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach. Hum Mutat. 2018;39:1641–49.
Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. 2017;19:1096–104.
Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, Topper S. Sources of discordance among germ-line variant classifications in ClinVar. Genet Med. 2017;19:1118–26.
Lee EM, Xu K, Mosbrook E, Links A, Guzman J, Adams DR, et al. Pharmacogenomic incidental findings in 308 families: the NIH undiagnosed diseases program experience. Genet Med. 2016;18:1303–7.
St Sauver JL, Bielinski SJ, Olson JE, Bell EJ, Mc Gree ME, Jacobson DJ, et al. Integrating pharmacogenomics into clinical practice: promise vs reality. Am J Med. 2016;129:1093–9.e1.
Crews KR, Gaedigk A, Dunnenberger HM, Leeder JS, Klein TE, Caudle KE, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharm Ther. 2014;95:376–82.
Hicks JK, Sangkuhl K, Swen JJ, Ellingrod VL, Müller DJ, Shimoda K, et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin Pharm Ther. 2017;102:37–44.
Acknowledgements
This work was supported by grants from Dijon University Hospital, the Regional Council of Burgundy Franche-Comté through the Plan d’Actions Régional pour l’Innovation (PARI 2016) and the European Union through the PO FEDER-FSE Bourgogne 2014/2020 programmes and the FEDER 2016. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Author contributions
CTR, JT, LF, CV, AC, EC, AP, CP, YD, MB, ML and CB designed the study. SN, JD, PK, ALB, EG, DL, NJM, PC, AMB, AV, AS, FTNM, CP, PV, LD and ML interpreted the exome data. CP, FTMT, JFD, CP, TJ and MC performed the molecular laboratory work. YD and ET performed the bioinformatics analysis. All the authors contributed to the writing and review of the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Thauvin-Robinet, C., Thevenon, J., Nambot, S. et al. Secondary actionable findings identified by exome sequencing: expected impact on the organisation of care from the study of 700 consecutive tests. Eur J Hum Genet 27, 1197–1214 (2019). https://doi.org/10.1038/s41431-019-0384-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0384-7
This article is cited by
-
An spanish study of secondary findings in families affected with mendelian disorders: choices, prevalence and family history
European Journal of Human Genetics (2023)
-
Exome sequencing allows detection of relevant pharmacogenetic variants in epileptic patients
The Pharmacogenomics Journal (2022)
-
An accessible insight into genetic findings for transplantation recipients with suspected genetic kidney disease
npj Genomic Medicine (2021)
